
ABSTRACT
Chimeric antigen receptor (CAR) T-cells have shown great promise in the treatment of B-cell malignancies. For acute myeloid leukemia (AML), however, the optimal target surface antigen has yet to be discovered. Alternatively, T-cell receptor (TCR)-redirected T-cells target intracellular antigens, marking a broader territory of available target antigens. Currently, adoptive TCR T-cell therapy uses peripheral blood lymphocytes for the introduction of a transgenic TCR. However, this can cause graft-versus-host disease, due to mispairing of introduced and endogenous TCR chains. Therefore, we started from hematopoietic stem and progenitor cells (HSPC), that do not express a TCR yet, isolated from healthy donors, patients in remission after chemotherapy and AML patients at diagnosis. Using the OP9-DL1 in vitro co-culture system and agonist selection, TCR-transduced HSPC develop into mature tumor antigen-specific T-cells with only one TCR. We show here that this approach is feasible with adult HSPC from clinically relevant sources, albeit with slower maturation and lower cell yield compared to cord blood HSPC. Moreover, cryopreservation of HSPC does not have an effect on cell numbers or functionality of the generated T-cells. In conclusion, we show here that it is feasible to generate TA-specific T-cells from HSPC from adult healthy donors and patients and we believe these T-cells could be of use as a very valuable form of patient-tailored T-cell immunotherapy.
Introduction
The last few years there’s been increasing evidence that T-cell-based immunotherapy is a successful treatment option for hematologic malignancies. Chimeric antigen receptor (CAR) T-cells are derived from a patient’s own immune cells and are able to recognize and target surface antigens independent of HLA. Most success has been achieved targeting CD19 in B-cell malignancies.Citation1,Citation2 For acute myeloid leukemia (AML), a suitable target surface antigen would ideally be expressed on leukemic blasts and also leukemic stem cells (LSC), which are often deemed responsible for relapse,Citation3 and not on indispensable normal hematopoietic cells. This target antigen, however, has yet to be discovered. (Pre)clinical studies targeting CD44v6,Citation4 CLL1,Citation5 FLT3,Citation6 LeY,Citation7 NKG2D ligands,Citation8,Citation9 CD33 and/or CD123Citation10-Citation12 frequently show on-target off-leukemia cytotoxicity. Another problem arising with targeting cell surface antigens is antigen downregulation, leading to escape and subsequent disease recurrence.Citation13,Citation14
Using a different approach, T-cell receptor (TCR)-modified T-cell therapy targets intracellular antigens which are often essential for cellular and/or oncogenic function and are therefore less prone to antigen escape. In TCR T-cell therapy, peripheral blood T-lymphocytes (PBL) are isolated and genetically engineered to express a transgenic TCR. Clinical trials with a TCR targeting Wilms’ tumor 1 (WT1), a tumor antigen overexpressed in a variety of cancers, including AML (in 80% of patientsCitation15), have already been initiated.Citation16–Citation19 In this approach, however, mispairing can occur between the endogenous and the introduced TCRα and TCRβ chains, leading to off-target toxicities.Citation20,Citation21 Moreover, intensive in vitro culturing of PBL potentially leads to exhausted TEFF/TEM T-cells with limited effector function and in vivo persistence,Citation22,Citation23 while it has been shown that less differentiated TN, TCM and especially TSCM are the most potent antitumor T-cells for T-cell immunotherapy.Citation24–Citation26
Multiple strategies are being explored to circumvent mispairing of TCR chains and extensive ex vivo culturing of T-cells. One strategy generates tumor antigen (TA)-specific T-cells by TCR transduction of induced pluripotent stem cells.Citation27 Another strategy, developed in our group, generates TA-specific T-cells with a single TCR and naive-like characteristics from TCR-transduced postnatal thymusCitation28 and cord blood (CB;Citation29) hematopoietic stem and progenitor cells (HSPC). This strategy is based on the OP9-DL1 in vitro co-culture system and agonist selection to induce T-cell differentiation from HSPC.
Here, we wanted to evaluate clinically more relevant HSPC sources in our model and generated functional, TA-specific T-cells from adult HSPC sources: healthy donors, patients in remission after chemotherapy, and AML patients at diagnosis. We show that this approach is feasible, both from healthy donors and patients, from fresh as well as cryopreserved samples, albeit with slower maturation and lower cell numbers as compared to cord blood HSPC.
Materials and methods
Isolation of human CD34+ cells
We collected cord blood, mobilized peripheral blood from patients undergoing autologous hematopoietic stem cell transplantation (HSCT) and from healthy donors for allogeneic HSCT, and peripheral blood, bone marrow, and leucapheresis from AML patients at diagnosis, with a CD34-negative AML. These samples were obtained and used following guidelines of the Medical Ethical Committee of the Ghent University Hospital. Informed consent was obtained in accordance with the Declaration of Helsinki.
Agonist peptide stimulation of HLA-A2 positive samples
Agonist peptide stimulation was carried out as described in Snauwaert et al.Citation28 In brief, cells were harvested from OP9-DL1 co-culture and seeded in tissue culture plates (BD Biosciences) in IMDM (Thermo Fisher Scientific, 12440053) supplemented with 10% fetal calf serum (FCS; Bovogen, SFBS-FR), 2 mM L-glutamine (Thermo Fisher Scientific, 25030–081), 100 IU/ml penicillin, and 100 IU/ml streptomycin (Thermo Fisher Scientific, 15140–122) (complete IMDM, cIMDM) with 10 ng/ml interleukin 7 (IL-7; R&D Systems, 207-IL-025) and 10 µg/ml relevant WT1126−134 agonist peptide (Anaspec by Eurogentec, custom peptide). Cells were harvested after 5–6 days and maturation was assessed by flow cytometry, as upregulation of CD27 and downregulation of CD1a. If necessary, cells were subjected to agonist peptide stimulation in the following rounds (maximum 3 rounds).
Cell-line dependent maturation of HLA-A2 negative samples
For HLA-A2 negative HSPC, maturation was obtained using co-culture with irradiated peptide-pulsed T2 cells. T2 cells were pulsed for 4 h with WT1126−134 peptide and irradiated (40 Gy). T-cell precursors were harvested from OP9-DL1 and seeded in tissue culture plates in cIMDM with 10 ng/ml IL-7. T2 cells were added at a 4/1 effector/target (E/T) ratio. Cells were harvested after 5–6 days and maturation was assessed by flow cytometry. If necessary, cells were stimulated with freshly peptide-pulsed and irradiated T2 cells in consecutive rounds (maximum 3 rounds).
Statistics
Statistical analyses were performed in Prism v5.01 (GraphPad Software), using statistical tests as indicated in figure legends. Results were considered statistically significant when P-value was less than 0.05.
Additional materials and methods are provided in Supplemental Data.
Results
CD34+ hematopoietic stem and progenitor cells from adult sources show slower in vitro maturation kinetics and less expansion compared to neonatal cord blood HSPC
We wanted to investigate the possibility of in vitro generation of TA-specific T-cells from clinically relevant HSPC sources, following the protocol previously described by our group.Citation28,Citation30 CD34+ HSPC were isolated from mobilized peripheral blood (mPB) samples from healthy donors (n = 13), mPB samples from patients in remission after chemotherapy (n = 16), and samples (bone marrow, peripheral blood, or leukapheresis) from AML patients at diagnosis, with CD34-negative AML (n = 13). Patient characteristics are shown in Supplementary Table S1.
We co-cultured isolated CD34+ HSPC from patient and healthy donor samples on OP9-DL1 cells until a significant population (50–80%) showed lymphoid lineage commitment, as evidenced by the combined surface expression of CD5 and CD7. With cord blood (CB) HSPC, this is generally at day 14 after initiation of co-culture. With adult HSPC sources (both patient and healthy), however, the kinetics to obtain a robust CD5+CD7+ population appeared to be slower ( and Supplementary Figure S1).
Figure 1. CD34+ HSPC from adult sources show slower in vitro maturation kinetics and less expansion compared to cord blood HSPC. (a) Culture protocol. Numbers indicate time (in days) of co-culture. Abbreviations: CB, cord blood; mPB, mobilized peripheral blood; BM, bone marrow; DP, double positive; SCF, stem cell factor; FLT3-L, FLT3 ligand; IL, interleukin; RV, retroviral. (b) Kinetics of expansion before transduction in OP9-DL1 co-cultures of HSPC from cord blood (n = 7), healthy donors (n = 12), patients in remission after chemotherapy (n = 15) and AML patients at diagnosis (n = 12). Mean ± s.d. is shown. T-cell committed progenitors in cord blood co-cultures were transduced at day 14, in co-cultures from adult HSPC at later timepoints (d19 or d24). (c) Relative cell numbers (i.e. cell numbers obtained when theoretically starting from a single CD34+ cell at day 0) at day 14 of co-cultures from cord blood (n = 7), healthy donor (n = 13), patient in remission after chemotherapy (n = 16) and AML patient at diagnosis (n = 13) HSPC. Values for individual samples and mean ± s.d. are shown. Mann–Whitney U test was used to assess statistical significance. P-value < 0.05 (*), P < 0.01 (**) and P < 0.001 (***)
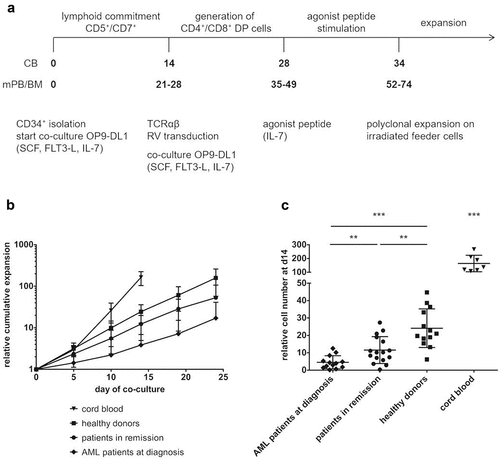
Healthy donor relative cell numbers were similar to those of CB at day of transduction (d14 for CB, d19 or 24 for healthy donors) (). Patient cells, however, showed lower overall expansion rates (). At day 14, significant differences in cell numbers could be observed between the different sample populations: fewer cells were generated from patient samples as compared to healthy donor samples, with patients in remission doing significantly better than AML patients at diagnosis (). At later timepoints (d19 and d24), the differences in relative cell numbers between adult sample populations remain significant (data not shown). Also, the relative cell yield at the end of the in vitro T-cell generation process, both before (Supplementary Figure S2A) and after (Supplementary Figure S2B) polyclonal feeder expansion, was significantly lower for co-cultures started from patient HSPC, as compared to healthy donor HSPC.
Since the healthy donors in our study were significantly younger compared to patients (Supplementary Table S1 and Supplementary Figure S3), we investigated if there was an effect of donor age on cell numbers. In our experiments, no significant correlation between age and cell numbers was observed at day 14 (Supplementary Figure S4) nor at later timepoints (data not shown).
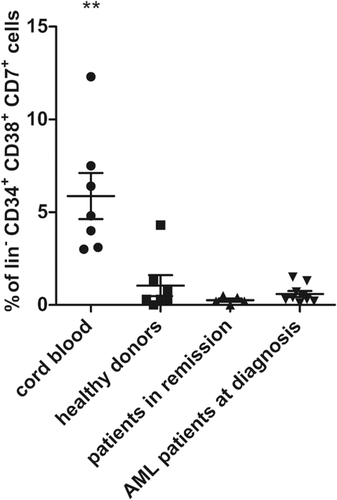
To further in-depth characterize the isolated CD34+ starting population, we analyzed the presence of different progenitor populations within the CD34+ population: hematopoietic stem cells (HSC, lin−CD34+CD38lo/-CD45RA−CD90+), multipotent progenitors (MPP, lin−CD34+CD38lo/-CD45RA−CD90−), multi-lymphoid progenitors (MLP, lin−CD34+CD38lo/-CD45RA+CD90−) and early T-progenitors (ETP, lin−CD34+CD38+CD7+).Citation31 Flow cytometric analysis revealed a higher percentage of early T-progenitors in CB HSPC as compared to adult HSPC (). No relevant differences in other progenitor fractions were observed. These data can explain faster maturation kinetics for CB, as compared to adult sample populations, but not differences between adult populations.
Figure 2. Presence of early T-progenitors at the start of co-cultures. Percentage of early T-progenitors (ETP, lin− CD34+ CD38+ CD7+) in CD34+ cells isolated at day 0 of co-cultures from cord blood (n = 7), healthy donors (n = 7), patients in remission (n = 5) and AML patients at diagnosis (n = 9). Individual samples, mean percentages per sample group and s.d. are shown. Mann–Whitney U test was used to assess statistical significance. P-value < 0.01 (**). Other differences were not significant
Multiple rounds of agonist peptide stimulation are needed to achieve the selection and maturation of HSPC from adult sources
When a large population of lymphoid-committed cells was present (50–80%), cells were transduced with a TA-specific TCR. At this point, intracellular CD3 was present. Here, we used the WT1-specific TCR, recognizing the HLA-A2-restricted peptide WT1126−134. Median transduction efficiency, measured 2 days after transduction, was 17.6% (range 10.2–27.7%), with donor-specific variations independent of sample group (data not shown).
Upon additional OP9-DL1 co-culture, TCR-transduced cells showed further T-lineage differentiation toward CD4+CD8+ double positive (DP) cells. At this stage, maturation to mature single positive T-cells can be obtained through agonist selection, as described by Snauwaert et al.Citation28 Briefly, for co-cultures from HLA-A2+ samples, the cognate peptide recognized by the transgenic TCR was added and cross-presented to induce maturation, as illustrated by transition from a CD27−CD1a+ to a CD27+CD1a− (mature) phenotype. For CB samples, the majority of the cells were CD1a− 6 days after agonist selection (). For co-cultures started from adult HSPC sources, however, only a small percentage was CD27+ and CD1a− at this timepoint. Therefore, cells were harvested and again subjected to agonist peptide stimulation. This process was repeated until approximately 30% of the cells were CD27+CD1a−. For co-cultures from healthy donor samples, 1 to 2 rounds of agonist peptide stimulation were needed to reach this point (). For co-cultures from patients in remission and from AML patients at diagnosis, up to 3 rounds were needed (). However, percentages of 60–70% CD27+CD1a− cells, as obtained in CB, were never reached in co-cultures from adult HSPC sources despite multiple rounds of agonist peptide stimulation.
Figure 3. Multiple rounds of agonist peptide stimulation are needed to achieve the selection and maturation of HSPC from adult sources. Maturation of TCR-transduced cells after one or more rounds of agonist peptide stimulation. Contour plots show CD1a (x-axis) and CD27 (y-axis) expression before agonist stimulation and after 1, 2 or 3 rounds of agonist stimulation for TCR-transduced cells in co-cultures from cord blood (a), healthy donors (b), patients in remission after chemotherapy (c) and AML patients at diagnosis (d). Gating on eGFP+ TCR-transduced cells. Numbers indicate percentages of cells in each quadrant. A representative sample from each sample group is shown
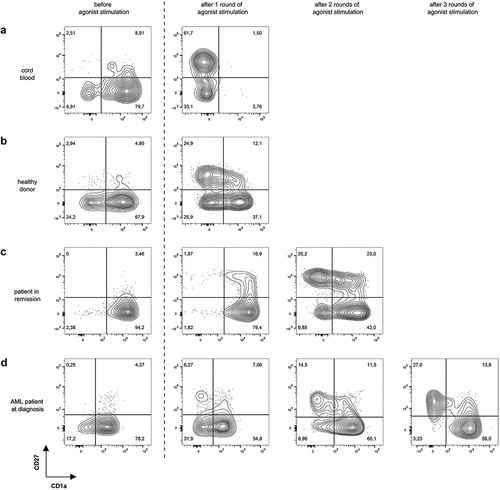
To make sure that the continued maturation we observed in adult samples with successive rounds of agonist peptide stimulation was due to upregulation of CD27 by remaining CD1a+CD27− DP cells, and not to proliferation of already present CD27+ cells, we sorted the remaining CD27− cells after the first round of agonist peptide stimulation and subjected these to another round of agonist peptide stimulation. These experiments confirmed additional maturation of CD27− cells with subsequent rounds of agonist peptide stimulation (data not shown).
Phenotype of in vitro generated TA-specific T-cells shows Tscm-like characteristics and largely lacks PD-1 expression
Some T-cell subsets are superior over others for use in adoptive T-cell immunotherapy. Especially more naive subsets (TN, TSCM, TCM) have been shown to exert a higher therapeutic efficiency.Citation24,Citation25 For this reason, we analyzed the phenotype of our in vitro generated T-cells, both after (every round of) agonist peptide stimulation and after polyclonal feeder expansion. T-cells were stained with various phenotypic markers (CD62L, CXCR3, CD56, NKG2D, CD57, CD95, CD127, CD45RA, CD45RO) and analyzed using flow cytometry. Since PD-1 was demonstrated to be a marker of T-cell exhaustion,Citation32 we also evaluated PD-1 expression.
Repetitive antigen stimulation is known to lead to a skewed effector memory (TEM) phenotype, with ensuing lower therapeutic efficacy.Citation33 Therefore, we first evaluated the effect of multiple rounds of agonist peptide stimulation on the phenotype of the cells. We compared cells that had been exposed to agonist peptide just once to cells that had undergone multiple rounds of agonist peptide stimulation. Altogether, we observed significant downregulation of CXCR4 and PD-1, and a trend toward upregulation of CXCR3 and CD45RO after repeated agonist peptide stimulation, a phenotype more skewed toward TEM/TTE (Supplementary Figure S5). Combining CD62L, CD45RA, CD95 and CXCR3 staining to identify T-cell subpopulations,Citation34 we observed a downregulation in the percentage of cells with a TN phenotype (CD45RA+ CD62L+ CXCR3− CD95−) and an upregulation in TTE cells (CD45RA+ CD62L− CXCR3− CD95+) after multiple rounds of agonist peptide stimulation (), consistent with the separate stain data displayed in Supplementary Figure S5.
Figure 4. Phenotype of in vitro generated T-cells after agonist peptide stimulation and after polyclonal feeder expansion. (a) Percentage of cells with a TN (CD45RA+ CD62L+ CXCR3− CD95−), TSCM (CD45RA+ CD62L+ CXCR3+ CD95+), TCM (CD45RA− CD62L+ CXCR3+ CD95+), TEM (CD45RA− CD62L− CXCR3− CD95+) and TTE (CD45RA+ CD62L− CXCR3− CD95+) phenotype before agonist peptide stimulation, and after 1, 2 or 3 rounds of agonist peptide stimulation. (b) Percentage of cells with a TN, TSCM, TCM, TEM, and TTE phenotype after polyclonal feeder expansion. For (a) and (b) T-cells were generated from HSPC from healthy donors (n = 3), patients in remission (n = 6) and AML patients at diagnosis (n = 3). Gating on eGFP+ TCR-transduced cells. Mean and s.d. are shown. (c) Percentage of cells positive for PD-1 expression after polyclonal feeder expansion. T-cells were generated from HSPC from healthy donors (n = 8), patients in remission after chemotherapy (n = 9) and AML patients at diagnosis (n = 6). Gating on eGFP+ TCR-transduced cells. Individual samples and mean ± s.d. are shown. Mann–Whitney U test was used to assess statistical significance. P-value < 0.05 (*)
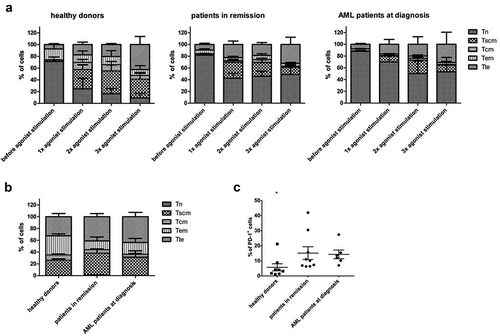
After agonist peptide-induced maturation, cells were polyclonally expanded on irradiated feeder cells, for a median of 12 days (range 10–14 days), to obtain phenotypically and functionally mature TA-specific T-cells. After polyclonal expansion, simultaneous staining with CD62L, CD45RA, CD95, and CXCR3 revealed an average of 33% of cells with a TSCM phenotype (CD45RA+ CD62L+ CXCR3+ CD95+)Citation34 (). More extensive staining further confirmed TSCM characteristics: CD57− CD45RO+ (Supplementary Figure S6).Citation34 CD62L expression was slightly downregulated as compared to expression after agonist peptide stimulation (50–60% and 80–90% of cells, respectively), which could be due to T-cell activation.Citation35 Moreover, cells upregulated expression of CXCR3, CD56 and NKG2D, additional markers of T-cell activation (cells were CD56−NKG2D− after agonist selection, data not shown).Citation28,Citation36,Citation37 Almost all cells showed CD95 expression, both before and after polyclonal feeder expansion (Supplementary Figure S5 and S6), which seems to be inherent to our in vitro OP9-DL1 co-cultures. CD127 (IL-7Rα) expression, another marker for TSCM,Citation34 could not be observed, both after agonist selection and after polyclonal expansion (data not shown and Supplementary Figure S6).
The T-cell exhaustion/activation marker PD-1,Citation32,Citation38 present after agonist selection, declined after polyclonal expansion in absence of the cognate antigen. Percentages of PD-1+ mature T-cells ranged from 1.1% to 42% (average 11.6%), marking donor-dependency (). Furthermore, T-cells generated from healthy donor HSPC had a significantly lower PD-1 expression compared to T-cells from patient HSPC. This difference in PD-1 expression between healthy donors and patients was independent of age (data not shown).
We compared the phenotype of our in vitro generated TA-specific T-cells to TCR-transduced PBL. For this, PBL were isolated, stimulated, transduced with a TA-specific TCR and polyclonally expanded on irradiated feeder cells. Staining with CD62L, CD45RA, CD95, and CXCR3 showed that the majority of the cells had a TN phenotype after isolation (Supplementary Figure S7). After stimulation, transduction, and subsequent polyclonal feeder expansion, 14.62% of TCR-transduced PBL showed a TSCM phenotype, 13.55% a TCM phenotype, 9.83% a TEM phenotype and 1.5% a TTE phenotype (Supplementary Figure S7).
In vitrogenerated TA-specific T-cells specifically recognize and kill TA-expressing cell lines
To investigate the functionality of in vitro generated WT1-specific T-cells from adult HSPC, T-cells were cultured with THP-1, a HLA-A2+WT1+ cell line, and JY cells (HLA-A2+WT1−), as a negative control. Interferon-gamma (IFNɣ) production after recognition of WT1+ target cells was measured through intracellular staining. In vitro generated T-cells, both from patient and healthy donor samples, specifically recognized WT1-presenting cells and, as a result, produced IFNɣ (median 12.85% of cells, range 4–48%) (). No significant difference could be observed in IFNɣ production between T-cells generated from healthy donor versus patient HSPC. Percentages of IFNɣ+ cells are in line with CB-derived T-cells (data not shown). We compared WT1-specific T-cells generated in vitro from adult HSPC, with WT1-specific T-cells generated from PBL. Whereas aspecific aCD3/aCD28 stimulation led to similar percentages of IFNɣ+ cells (median 42.93% of cells, range 10.95–82.3% for HSPC and median 46.02% of cells, range 26.5 – 56% for PBL), specific recognition of WT1+ target cells was significantly lower in PBL-derived T-cells (median 1.51% of cells, range 0.85–2.2%) ().
Figure 5. In vitro generated TA-specific T-cells specifically recognize and kill TA-expressing cell lines. (a) Intracellular staining of T-cells for interferon-gamma (IFNg) after co-culture with THP-1 (HLA-A2+ WT1+) or JY (HLA-A2+ WT1−) cells. Culture medium was used as a negative control, stimulation with aCD3/aCD28 as a positive control. Effector/target ratio 1/2. Gating on eGFP+ TCR-transduced cells. T-cells generated from HSPC from healthy donors (n = 4), patients in remission after chemotherapy (n = 5), AML patients at diagnosis (n = 4) and PBL (n = 6). (b) Percentage specific lysis determined via 4-h 51chromium release assay after co-culture of T-cells with T2 cells pulsed with relevant WT1 or irrelevant influenza (INF) peptide (10 µg/ml), HL-60-A2 (HLA-A2+ WT1+), THP-1, or JY cells. Effector/target ratio 10/1. T-cells generated from HSPC from healthy donors (n = 6), patients in remission after chemotherapy (n = 7), AML patients at diagnosis (n = 8) and PBL (n = 6). For (a) and (b) mean and s.d. are shown. Kruskal–Wallis test was used to determine statistical significance between different HSPC sample groups. Differences were not significant. Mann–Whitney U test was used to determine statistical significance for between-group comparisons for HSPC- and PBL-derived T-cells. P-value < 0.05 (*) and P < 0.01 (**)
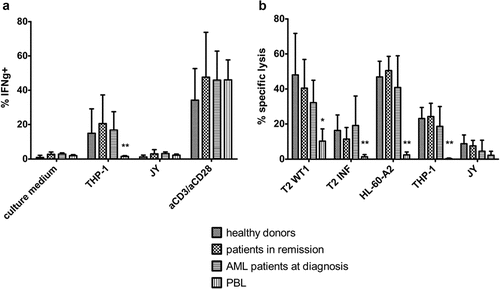
Furthermore, specific lysis of target cell lines was determined using a 51chromium release assay. For this, T2 cells were pulsed with relevant WT1 or irrelevant influenza peptide, and the HL-60-A2 cell line was used as an additional HLA-A2+WT1+ target (qPCR showed higher WT1 expression on HL-60-A2 compared to THP-1, data not shown). An average of 35.7% (range 5.1–87.8%) of HLA-A2+WT1+ cells were killed, whereas HLA-A2+WT1− cells were killed only to a minimal extent (). T-cells generated from HSPC from AML patients at diagnosis tended to lyse a smaller percentage of target cells, but differences were not significant. PBL-derived WT1-specific T-cells were able to kill only small percentages of HLA-A2+WT1+ target cells (4.34% of cells on average) ().
It has been shown that exposing maturing T-cells multiple times to antigen, as we did with the repeated agonist peptide stimulation, could lead to exhaustion and diminished functionality.Citation39,Citation40 Therefore, we evaluated the effect of multiple agonist peptide stimulations, in reference to only one stimulation, on the functionality of resulting T-cells. While repetitive antigen stimulation did seem to have a (minor) effect on phenotype, no significant differences could be found regarding the functionality of the cells. IFNɣ production after 3 rounds of agonist peptide stimulation appeared lower, but differences were not significant (Supplementary Figure S8A). 51Chromium release assays revealed no differences in lysis of HLA-A2+WT1+ targets (Supplementary Figure S8B).
Cryopreservation has a negligible effect on maturation kinetics and functionality of in vitro generated TA-specific T-cells
The CD34+ HSPC pool of patients can be damaged by chemotherapy and other treatments, rendering a stem cell harvest difficult. Therefore, we investigated the possibility of collecting HSPC beforehand at a time of remission, and cryopreserving them for longer periods of time, before generating TA-specific T-cells for therapy. Also, cryopreserved healthy donor HSPC could be used for the generation of off-the-shelf T-cells for cancer therapy, and for donor-derived adoptive T-cell therapy after haploidentical HSCT. We evaluated the effect of cryopreservation both on maturation kinetics of T-cell precursors in OP9-DL1 co-cultures, and functionality of the resulting mature T-cells.
After sample collection and density gradient centrifugation, part of the cells was frozen and part of the cells used for further CD34+ HSPC isolation and ‘fresh’ OP9-DL1 co-culture. Cryopreserved cells were thawed, CD34+ cells were isolated and cultured on OP9-DL1. We compared the relative cumulative expansion of fresh and frozen HSPC from CB, healthy donors, and patients (both in remission and at diagnosis) (Supplementary Figure S9A). At day 14, no notable differences in relative cell numbers could be found between fresh and frozen HSPC (). Also, relative cell yield of mature T-cells at the end of co-cultures, both before (Supplementary Figure S9B) and after (Supplementary Figure S9C) polyclonal feeder expansion, was similar for T-cells generated from fresh and frozen HSPC.
Figure 6. Cryopreservation has a negligible effect on maturation kinetics and the functionality of in vitro generated TA-specific T-cells. (a) Relative cell numbers at day 14 of co-cultures (day at which cord blood progenitors were transduced) from fresh and cryopreserved (frozen) HSPC from cord blood (n = 6), healthy donors (n = 6), patients in remission after chemotherapy (n = 7) and AML patients at diagnosis (n = 6). Individual fresh and paired frozen samples are shown. (b) Percentage specific lysis determined via 4-h 51chromium release assay after co-culture of T-cells with T2 cells pulsed with relevant WT1 or irrelevant influenza (INF) peptide (10 µg/ml), HL-60-A2 (HLA-A2+ WT1+), THP-1 (HLA-A2+ WT1+) or JY (HLA-A2+ WT1−) cells. Effector/target ratio 5/1. T-cells generated from fresh and cryopreserved (frozen) HSPC (n = 11). Results from different sample groups (healthy donors, patients in remission and AML patients at diagnosis) were pooled. Mean and s.d. are shown. Wilcoxon matched-pairs signed-rank test was used to assess statistical significance. Differences were not significant
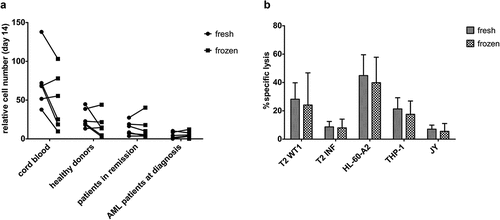
At the end of the in vitro culture protocol, functionality experiments revealed significantly higher IFNɣ production after HLA-A2+WT1+ target recognition by T-cells generated from fresh samples as compared to cryopreserved samples (Supplementary Figure S10). However, lysis of target cell lines appeared unaffected by cryopreservation ().
Mature TA-specific T-cells can also be generated from HSPC of HLA-A2 negative healthy donors
To try and circumvent HLA restrictions incurred by the use of a transgenic TCR, we wanted to investigate whether it would be possible to generate HLA-A2-restricted TA-specific T-cells to administer to a HLA-A2+ patient, starting from CD34+ cells from HLA-A2− donors. Agonist peptide cross-presentation, to obtain maturation from CD1a+CD27− TCR-transduced DP cells to CD1a−CD27+ SP cells,Citation28 is not possible with HLA-A2− samples due to HLA-restriction of the TCR. Snauwaert et al. demonstrated that the addition of HLA-A2+ dendritic cells (DC) together with the agonist peptide to HLA-A2− co-cultures results in T-cell maturation.Citation28 To optimize this protocol, we excluded the possibility of other peptides being presented on the surface of the DC, by using the transporter associated with antigen processing (TAP)-deficient HLA-A2+ T2 cell line as antigen-presenting cells. T2 cells fail to present endogenous peptides and can be loaded with exogenously administered peptides. At the DP stage, we added the T2 cell line pulsed with the agonist peptide in co-cultures from HLA-A2− HSPC. Subsequent downregulation of CD1a expression could be observed, in accordance with our observations for HLA-A2+ DP cells (data not shown). However, upregulation of CD27 does not occur, probably due to the binding of CD27 on T-cell precursors with CD70 expressed by T2 cells (data not shown).
Although phenotypic analysis of mature TA-specific T-cells demonstrated significantly higher CD62L expression in HLA-A2− as compared to HLA-A2+ donor-derived T-cells, overall phenotype remained similar (Supplementary Figure S11A).
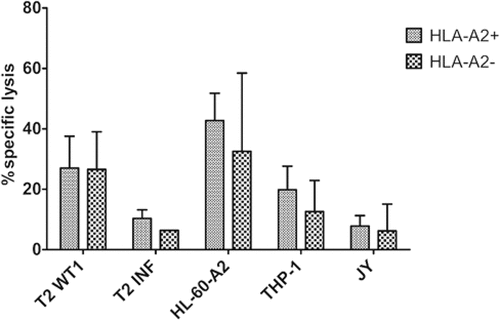
Comparing the functionality of TA-specific T-cells generated from HLA-A2+ and HLA-A2− HSPC, we found no significant differences in IFNγ production after recognition of HLA-A2+WT1+ target cells (Supplementary Figure S11B). Also, 51chromium release assays showed no difference in killing of target cell lines (), indicating that it is possible to generate functional TA-specific T-cells, recognizing the TA in a HLA-A2+ context, from both HLA-A2+ and HLA-A2− donors.
Figure 7. Functional TA-specific T-cells can also be generated from HSPC from HLA-A2 negative donors. Percentage specific lysis determined via 4-h 51chromium release assay after co-culture of T-cells with T2 cells pulsed with relevant WT1 or irrelevant influenza (INF) peptide (10 µg/ml), HL-60-A2 (HLA-A2+ WT1+), THP-1 (HLA-A2+ WT1+) or JY (HLA-A2+ WT1−) cells. Effector/target ratio 5/1. Mean and s.d. are shown. T-cells generated from HLA-A2+ (n = 5) and HLA-A2− (n = 4) HSPC. Results from different sample groups (healthy donors, patients in remission and AML patients at diagnosis) were pooled. Mann-Whitney U test was used to assess statistical significance. Differences were not significant
Discussion
We have shown that it is feasible to generate functional TA-specific T-cells from clinically relevant stem cell sources, using the OP9-DL1 in vitro co-culture system and agonist selection.Citation28 T-cell differentiation kinetics from adult HSPC sources were slower compared to the kinetics of CB samples. These results are in line with a publication by Offner and colleagues, who found that bone marrow (BM)-derived HSPC from older (>40 years) donors had a lower capacity to reach the CD4+CD8+ double-positive stage of T-cell differentiation in fetal thymic organ cultures, as compared to HSPC from younger donors.Citation41 This indicates an impact of donor age on T-cell generation capacity (in vitro). We did, however, not find a significant correlation between age and cell numbers. A possible explanation could be the limited age range within sample groups, with healthy donors being mostly younger (<40 years) and patients being older (>40 years). Healthy donor-derived T-cells possessing only one TA-specific TCR could be used in the context of HSCT, even after haploidentical HSCT, thereby avoiding graft-versus-host disease (GVHD) caused by a partial HLA mismatch.Citation42
In patients, chemotherapy has been shown to have a detrimental effect on T-cells. Total CD8+ lymphocyte counts restore within three to 6 months after cessation of cytotoxic therapy, whereas CD4+ T-cells suffer a prolonged lymphopenia, resulting in T-cell subset inversed ratio and repertoire skewing.Citation43 As T-cell-based immunotherapy is currently positioned for high-risk patients after several lines of (chemo)therapy, we wanted to evaluate the generation of TA-specific T-cells from HSPC isolated from patients in remission. We show that the heavy pre-treatment of these patients resulted in slower T-cell differentiation of isolated HSPC in OP9-DL1 co-cultures, and the need for multiple rounds of agonist peptide stimulation to reach a phenotypically mature CD1a−CD27+ population. Both observations indicate a diminished T-cell potential of HSPC following cytotoxic therapy.
Nevertheless, we could show the generation of functional, mature T-cells from HSPC from patients in remission, albeit with lower cell numbers as compared to younger healthy sources. Combining cell yield data (before feeder expansion) with the number of CD34+ cells that could be isolated from patients or healthy donors, the minimum number of T-cells that we can generate is 2.2 x 105/kg from AML patients (up to 1.99 x 108/kg), 61.6 × 106 from patients in remission (up to 1.92 x 109/kg) and 6.21 × 108 from healthy donors (up to 1.25 x 1011/kg). With CAR T-cells, as low as 1.5 × 105 cells per kilogram of bodyweight have been injected into patients, showing expansion, persistence and disease eradication,Citation44 suggesting our final cell numbers may be sufficient.
In acute myeloid leukemia, a complete remission can be achieved in roughly 50% of patients with the classical 7 + 3 chemotherapy regimen,Citation45 and even in this group the chance of relapse is high (40 – 75%). Current treatment options for relapsed or refractory AML only offer a bridge-to-transplantation since no other curative option has been found yet. In primary or secondary refractory AML patients, and patients at risk for relapse, T-cell-based immunotherapy could be a new treatment option, potentially even replacing HSCT. In this setting, HSPC needs to be isolated from blood or bone marrow with a high AML blast percentage. This is similar to CD19 CAR T-cell therapy in refractory ALL patients, where T-cells need to be isolated from peripheral blood containing high numbers of malignant blasts.Citation46 Therefore, we evaluated the feasibility to generate TA-specific T-cells starting with HSPC from leucapheresis, PB, or BM samples from AML patients at diagnosis, who, like refractory patients, have a high number of circulating leukemic cells. We selected AML patients with a CD34-negative AML, as LSC are predominantly CD34+.Citation47,Citation48 However, as there has been controversy about the LSC immunophenotype,Citation49 it might be necessary to perform extensive genetic analyses before reintroducing our T-cells back into the patient, to make sure no leukemic cells are present. On the other hand, a study by Ruella et al.Citation50 showed that in CD19 CAR-T treatment of B-ALL, presence of rare leukemic blasts (<0.01%) at the time of infusion did not correlate with relapse rate or time to relapse, indicating that presence of a small amount of leukemic cells in the infusion product not necessarily leads to relapse.
Patient cells usually need to be cryopreserved for practical reasons in the course of treatment (e.g. before myeloablative chemotherapy). Therefore, we investigated the effect of cryopreservation and thawing on the in vitro T-cell generation process. In stem cell transplantation clinical practice, stem cells are often cryopreserved (CB and autologous stem cell products) and thawed before infusion. The reconstitutive capacity of HSPC, cryopreserved for more than a decade, has been evidenced by in vitro assays for CBCitation51 and BMCitation52 HSPC. More extensive in vivo assays in immunodeficient mice demonstrated repopulation features of CB HSPC cryopreserved for up to 23.5 years, with multi-lineage engraftment in mice at engraftment levels comparable with those reported for fresh HSPC.Citation51,Citation53 Patient studies investigating the effect of cryopreservation of HSPC from different sources (CB, mPB, and BM) suggest that cryopreserved cells are not inferior to freshly collected cells at different outcomes measured, including engraftment rates, overall survival and GVHD.Citation54,Citation55 Our study showed small but irrelevant differences between fresh and cryopreserved samples. These results suggest it would be possible to harvest stem cells from patients before exposing them to detrimental effects of chemotherapy, and cryopreserving these HSPC for later in vitro T-cell generation. In vitro generated TA-specific T-cells can then be used as a treatment strategy to eliminate LSC and thereby minimal residual disease in a patient in remission.
Even in the current CAR-T era, research into TCR-directed therapy is indispensable. TCR, as opposed to CAR, recognize intracellular antigens, making them able to recognize a larger array of potential targets, which are also less prone to antigen escape.Citation13 TCR targets can be tumor-specific antigens, such as mutated (neo)antigens exclusively expressed by tumor cells or antigens re-expressed after embryogenesis, or tumor-associated antigens (TAA). TAA, such as WT1, are self-antigens overexpressed by tumor cells. TAA-specific TCR can be induced in an allo-reactive setting,Citation56 circumventing negative selection processes in the thymus. Moreover, affinity-enhancement of TCR has made it possible to optimize the affinity of self-antigen reactive TCR,Citation57,Citation58 since receptor affinities for self-antigen are generally low, even in an allo-setting. Direct comparisons of TCR and CAR with similar affinities have revealed a greater sensitivity in favor of TCR.Citation59,Citation60
The benefits of using the OP9-DL1 co-culture system to generate TCR-transduced T-cells are two-fold. On the one hand, we generate T-cells with a single TA-specific TCR and no endogenous TCR, circumventing the possibility of inducing toxicity, as seen when introducing a transgenic TCR in PBL that already express an endogenous TCR.Citation20,Citation21 Furthermore, we show here that HSPC-derived TA-specific T-cells are more functional against TA-expressing target cells, when compared to PBL-derived TA-specific T-cells. This confirms our previous data, where we have shown that this is due to higher TA-specific TCR expression on transduced HSPC.Citation28 On the other hand, we generate T-cells with a favorable TSCM phenotype, believed to be of optimal functionality and longevity in vivo, as opposed to end-stage phenotype T-cells generated with extensive in vitro culture protocols.Citation22,Citation61 On top of that, PD-1 expression is low, even when cells are repeatedly stimulated with the agonist peptide. This is in contrast to the article by Bucks et al., describing the upregulation of PD-1 expression after chronic antigen exposure.Citation39 In our model, repeated stimulation with the cognate peptide at the CD4+CD8+ DP stage of T-cell maturation leads to agonist selection instead of exhaustion.Citation28 As we expect our T-cells to expand in vivo after injection, we opt to inject them before in vitro feeder expansion, to avoid extensive culture and skewing toward TEM/TTE phenotypes. Since functionality is not enhanced by multiple rounds of agonist peptide stimulation (before polyclonal feeder expansion), we would opt to do one round of agonist peptide stimulation before injection, as these cells show a favorable phenotype resembling mostly TN/TSCM.
As our in vitro culture protocol is a universal protocol, the utilized TCR is interchangeable and not restricted to a certain target. Hence, any high-affinity TCR can be isolated or designed and used for the transduction of T-cell precursors and subsequent generation of TA-specific T-cells. Moreover, our group has previously shown that it is also possible to generate CAR T-cells from HSPC using the OP9-DL1 co-culture system.Citation29 The resulting CAR+ T-cells were CD3−TCRαβ−, and the endogenous TCR loci not rearranged, excluding the possibility of alloreactivity. In the event that a suitable target surface antigen for AML is found, we can use the same clinically relevant HSPC sources as mentioned in this paper to generate CAR T-cells expressing only the CAR and no endogenous TCR.
We have shown here that it is feasible to generate TA-specific T-cells from HSPC from adult healthy donors and patients. We believe these T-cells could be of use as a very valuable form of patient-tailored T-cell immunotherapy, after being submitted to further extensive preclinical analyses.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download ()Supplemental Material
Download ()Supplemental Material
Download ()Acknowledgments
The authors would like to thank P. Devreker, S. De Smet and V. Van De Steene of the Hematopoietic Stem Cell Bank, Dr. C. Matthys of the Cord Blood Bank, and all doctors of the Hematology department (all from Ghent University Hospital) for providing samples. We are indebted to Dr. B. Descamps and Prof. Dr. C. Vanhove of the Infinity lab of Ghent University for help with irradiation of cells, S. Vermaut for help with flow cytometry and cell sorting and T. Geudens for the artwork.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, Ghobadi A, Budde LE, Bot A, Rossi JM, et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285–12. doi:10.1016/j.ymthe.2016.10.020.
- Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, et al. CD19 CAR-T cells of defined CD4(+): CD8(+) composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123–2138. doi:10.1172/JCI85309.
- Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, Medeiros JJF, Rao-Bhatia A, Jaciw-Zurakowsky I, Marke R, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature. 2017;547(7661):104-+. doi:10.1038/nature22993.
- Casucci M, Di Robilant BN, Falcone L, Camisa B, Norelli M, Genovese P, Gentner B, Gullotta F, Ponzoni M, Bernardi M, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013;122(20):3461–3472. doi:10.1182/blood-2013-04-493361.
- Wang J, Chen S, Xiao W, Li W, Wang L, Yang S, Wang W, Xu L, Liao S, Liu W, et al. CAR-T cells targeting CLL-1 as an approach to treat acute myeloid leukemia. J Hematol Oncol. 2018;11(1):7. doi:10.1186/s13045-017-0553-5.
- Chien CD, Sauter CT, Ishii K, Nguyen SM, Shen F, Tasian SK, Chen W, Dimitrov DS, Fry TJ. Preclinical development of FLT3-redirected chimeric antigen receptor T cell immunotherapy for acute myeloid leukemia. Blood. 2016;128(22):1072. doi:10.1182/blood.V128.22.1072.1072.
- Peinert S, Prince HM, Guru PM, Kershaw MH, Smyth MJ, Trapani JA, Gambell P, Harrison S, Scott AM, Smyth FE, et al. Gene-modified T cells as immunotherapy for multiple myeloma and acute myeloid leukemia expressing the Lewis Y antigen. Gene Ther. 2010;17(5):678–686. doi:10.1038/gt.2010.21.
- Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73(6):1777–1786. doi:10.1158/0008-5472.CAN-12-3558.
- VanSeggelen H, Hammill JA, Dvorkin-Gheva A, Tantalo DG, Kwiecien JM, Denisova GF, Rabinovich B, Wan Y, Bramson JL. T cells engineered with chimeric antigen receptors targeting NKG2D ligands display lethal toxicity in mice. Mol Ther. 2015;23(10):1600–1610. doi:10.1038/mt.2015.119.
- Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJD, Scholler J, Song D, Porter DL, Carroll M, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29(8):1637–1647. doi:10.1038/leu.2015.52.
- Mardiros A, Dos Santos C, McDonald T, Brown CE, Wang XL, Budde LE, Hoffman L, Aguilar B, Chang W-C, Bretzlaff W, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122(18):3138–3148. doi:10.1182/blood-2012-12-474056.
- Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, Lassailly F, Tettamanti S, Spinelli O, Biondi A, Biagi E, Bonnet D. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. 2014;28(8):1596–1605. doi:10.1038/leu.2014.62.
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi:10.1056/NEJMoa1407222.
- Topp MS, Gokbuget N, Zugmaier G, Degenhard E, Goebeler ME, Klinger M, Neumann SA, Horst HA, Raff T, Viardot A, et al. Long-term follow-up of hematologic relapse-free survival in a phase 2 study of blinatumomab in patients with MRD in B-lineage ALL. Blood. 2012;120(26):5185–5187. doi:10.1182/blood-2012-07-441030.
- Rosenfeld C, Cheever MA, Gaiger A. WT1 in acute leukemia, chronic myelogenous leukemia and myelodysplastic syndrome: therapeutic potential of WT1 targeted therapies. Leukemia. 2003;17(7):1301–1312. doi:10.1038/sj.leu.2402988.
- Chapuis AG, Egan DN, Bar M, Schmitt TM, McAfee MS, Paulson KG, Voillet V, Gottardo R, Ragnarsson GB, Bleakley M, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med. 2019;25(7):1064–1072. doi:10.1038/s41591-019-0472-9.
- Autologous T cells with or without cyclophosphamide and fludarabine in treating patients with recurrent or persistent advanced ovarian epithelial cancer, primary peritoneal cavity cancer, or fallopian tube cancer (fludarabine treatment closed as of 12/01/2009). Accessed on 03/06/2019. https://ClinicalTrials.gov/show/NCT00562640
- Genetically modified T cells in treating patients with stage III-IV non-small cell lung cancer or mesothelioma. Accessed on 03/06/2019. https://ClinicalTrials.gov/show/NCT02408016
- A phase I/II study of gene-modified WT1 TCR therapy in MDS & AML patients. Accessed on 12/06/2018. Available from: https://ClinicalTrials.gov/show/NCT02550535.
- Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, Kaiser ADM, Pouw N, Debets R, Kieback E, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med. 2010;16(5):565–U98. doi:10.1038/nm.2128.
- van Loenen MM, de Boer R, Amir AL, Hagedoorn RS, Volbeda GL, Willemze R, van Rood JJ, Falkenburg JHF, Heemskerk MHM. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc Natl Acad Sci. 2010;107(24):10972–10977. doi:10.1073/pnas.1005802107.
- Janelle V, Carli C, Taillefer J, Orio J, Delisle JS. Defining novel parameters for the optimal priming and expansion of minor histocompatibility antigen-specific T cells in culture. J Transl Med. 2015;13:13. doi:10.1186/s12967-015-0495-z.
- Kagoya Y, Nakatsugawa M, Ochi T, Cen YC, Guo TX, Anczurowski M, Saso K, Butler MO, Hirano N. Transient stimulation expands superior antitumor T cells for adoptive therapy. JCI Insight. 2017;2(2):13. doi:10.1172/jci.insight.89580.
- Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu ZY, Finkelstein SE, Theoret MR, Rosenberg SA, Restifo NP. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8(+) T cells. J Clin Invest. 2005;115(6):1616–1626. doi:10.1172/JCI24480.
- Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17(10):1290–U325. doi:10.1038/nm.2446.
- Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12(10):671–684. doi:10.1038/nrc3322.
- Minagawa A, Yoshikawa T, Yasukawa M, Hotta A, Kunitomo M, Iriguchi S, Takiguchi M, Kassai Y, Imai E, Yasui Y, et al. Enhancing T Cell Receptor Stability in Rejuvenated iPSC-Derived T Cells Improves Their Use in Cancer Immunotherapy. Cell Stem Cell. 2018;23(6):850-+. doi:10.1016/j.stem.2018.10.005.
- Snauwaert S, Verstichel G, Bonte S, Goetgeluk G, Vanhee S, Van Caeneghem Y, De Mulder K, Heirman C, Stauss H, Heemskerk MHM, et al. In vitro generation of mature, naive antigen-specific CD8 (+) T cells with a single T-cell receptor by agonist selection. Leukemia. 2014;28(4):830–841. doi:10.1038/leu.2013.285.
- Van Caeneghem Y, De Munter S, Tieppo P, Goetgeluk G, Weening K, Verstichel G, Bonte S, Taghon T, Leclercq G, Kerre T, et al. Antigen receptor-redirected T cells derived from hematopoietic precursor cells lack expression of the endogenous TCR/CD3 receptor and exhibit specific antitumor capacities. OncoImmunology. 2017;6(3):14. doi:10.1080/2162402X.2017.1283460.
- Van Coppernolle S, Verstichel G, Timmermans F, Velghe I, Vermijlen D, De Smedt M, Leclercq G, Plum J, Taghon T, Vandekerckhove B, et al. Functionally mature CD4 and CD8 TCRαβ cells are generated in OP9-DL1 cultures from human CD34+ hematopoietic cells. J Immunol. 2009;183(8):4859–4870. doi:10.4049/jimmunol.0900714.
- Scala S, Basso-Ricci L, Dionisio F, Pellin D, Giannelli S, Salerio FA, Leonardelli L, Cicalese MP, Ferrua F, Aiuti A, et al. Dynamics of genetically engineered hematopoietic stem and progenitor cells after autologous transplantation in humans. Nat Med. 2018;24(11):1683-+. doi:10.1038/s41591-018-0195-3.
- Barber DL, Wherry EJ, Masopust D, Zhu BG, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–687. doi:10.1038/nature04444.
- Wirth TC, Xue -H-H, Rai D, Sabel JT, Bair T, Harty JT, Badovinac VP. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8+ T cell differentiation. Immunity. 2010;33(1):128–140. doi:10.1016/j.immuni.2010.06.014.
- Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T memory stem cells in health and disease. Nat Med. 2017;23:18. doi:10.1038/nm.4241.
- Yang S, Liu F, Wang QJ, Rosenberg SA, Morgan RA, Teague RM. The shedding of CD62L (L-selectin) regulates the acquisition of lytic activity in human tumor reactive T lymphocytes. PLoS One. 2011;6(7):e22560. doi:10.1371/journal.pone.0022560.
- Kelly-Rogers J, Madrigal-Estebas L, O’Connor T, Doherty DG. Activation-induced expression of CD56 by T cells is associated with a reprogramming of cytolytic activity and cytokine secretion profile in vitro. Hum Immunol. 2006;67(11):863–873. doi:10.1016/j.humimm.2006.08.292.
- Verneris MR, Karimi M, Baker J, Jayaswal A, Negrin RS. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood. 2004;103(8):3065–3072. doi:10.1182/blood-2003-06-2125.
- Chikuma S, Terawaki S, Hayashi T, Nabeshima R, Yoshida T, Shibayama S, Okazaki T, Honjo T. PD-1-mediated suppression of IL-2 production induces CD8+ T cell anergy in vivo. The Journal of Immunology. 2009;182(11):6682–6689. doi:10.4049/jimmunol.0900080.
- Bucks CM, Norton JA, Boesteanu AC, Mueller YM, Katsikis PD. Chronic antigen stimulation alone is sufficient to drive CD8(+) T cell exhaustion. J Immunol. 2009;182(11):6697–6708. doi:10.4049/jimmunol.0800997.
- Lang KS, Recher M, Navarini AA, Harris NL, Lohning M, Junt T, Probst H, Hengartner H, Zinkernagel R. Inverse correlation between IL-7 receptor expression and CD8 T cell exhaustion during persistent antigen stimulation. Eur J Immunol. 2005;35(3):738–745. doi:10.1002/eji.200425828.
- Offner F, Kerre T, De Smedt M, Plum J. Bone marrow CD34 cells generate fewer T cells in vitro with increasing age and following chemotherapy. Br J Haematol. 1999;104(4):801–808. doi:10.1046/j.1365-2141.1999.01265.x.
- Huang XJ, Liu DH, Liu KY, Xu LP, Chen H, Han W. Donor lymphocyte infusion for the treatment of leukemia relapse after HLA-mismatched/haploidentical T-cell-replete hematopoietic stem cell transplantation. Haematol-Hematol J. 2007;92(3):414–417. doi:10.3324/haematol.10570.
- Mackall CL, Fleisher TA, Brown MR, Andrich MP, Chen CC, Feuerstein IM, Magrath IT, Wexler LH, Dimitrov DS, Gress RE, et al. Distinctions between CD8(+) and CD4(+) T-cell regenerative pathways result in prolonged T-cell subset imbalance after intensive chemotherapy. Blood. 1997;89(10):3700–3707. doi:10.1182/blood.V89.10.3700.
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi:10.1056/NEJMoa1103849.
- Dombret H, Gardin C. An update of current treatments for adult acute myeloid leukemia. Blood. 2016;127(1):53–61. doi:10.1182/blood-2015-08-604520.
- Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–4828. doi:10.1182/blood-2011-04-348540.
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645. doi:10.1038/367645a0.
- Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, Nakamura R, Tanaka T, Tomiyama H, Saito N, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25:1315. doi:10.1038/nbt1350.
- Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, Poeppl A, Ling V, Beyene J, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17:1086. doi:10.1038/nm.2415.
- Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, Klichinsky M, Shestova O, Patel PR, Kulikovskaya I, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med. 2018;24(10):1499–1503. doi:10.1038/s41591-018-0201-9.
- Broxmeyer HE, Srour EF, Hangoc G, Cooper S, Anderson SA, Bodine DM. High-efficiency recovery of functional hematopoietic progenitor and stem cells from human cord blood cryopreserved for 15 years. Proc Natl Acad Sci U S A. 2003;100(2):645–650. doi:10.1073/pnas.0237086100.
- Donnenberg AD, Koch EK, Griffin DL, Stanczak HM, Kiss JE, Carlos TM, BuchBarker DM, Yeager AM. Viability of cryopreserved BM progenitor cells stored for more than a decade. Cytotherapy. 2002;4(2):157–163. doi:10.1080/146532402317381866.
- Broxmeyer HE, Lee MR, Hangoc G, Cooper S, Prasain N, Kim YJ, Mallett C, Ye Z, Witting S, Cornetta K, et al. Hematopoietic stem/progenitor cells, generation of induced pluripotent stem cells, and isolation of endothelial progenitors from 21- to 23.5-year cryopreserved cord blood. Blood. 2011;117(18):4773–4777. doi:10.1182/blood-2011-01-330514.
- Stockschlader M, Hassan HT, Krog C, Kruger W, Loliger C, Horstman M, ALTNODER M, CLAUSEN J, GRIMM J, KABISCH H, et al. Long-term follow-up of leukaemia patients after related cryopreserved allogeneic bone marrow transplantation. Br J Haematol. 1997;96(2):382–386. doi:10.1046/j.1365-2141.1997.d01-2032.x.
- Kim DH, Jamal N, Saragosa R, Loach D, Wright J, Gupta V, Kuruvilla J, Lipton JH, Minden M, Messner HA, et al. Similar outcomes of cryopreserved allogeneic peripheral stem cell transplants (PBSCT) compared to fresh allografts. Biol Blood Marrow Transplant. 2007;13(10):1233–1243. doi:10.1016/j.bbmt.2007.07.003.
- Gao L, Bellantuono I, Elsasser A, Marley SB, Gordon MY, Goldman JM, Stauss HJ. Selective elimination of leukemic CD34(+) progenitor cells by cytotoxic T lymphocytes specific for WT1. Blood. 2000;95(7):2198–2203. doi:10.1182/blood.V95.7.2198.
- Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, Dunn S, Liddy N, Jacob J, Jakobsen BK, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol. 2005;23(3):349–354. doi:10.1038/nbt1070.
- Holler PD, Holman PO, Shusta EV, O’Herrin S, Wittrup KD, Kranz DM. In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc Natl Acad Sci U S A. 2000;97(10):5387–5392. doi:10.1073/pnas.080078297.
- Harris DT, Hager MV, Smith SN, Cai Q, Stone JD, Kruger P, Lever M, Dushek O, Schmitt TM, Greenberg PD, et al. Comparison of T cell activities mediated by human TCRs and CARs that use the same recognition domains. The Journal of Immunology. 2018;200(3):1088–1100. doi:10.4049/jimmunol.1700236.
- Stone JD, Harris DT, Soto CM, Chervin AS, Aggen DH, Roy EJ, Kranz DM. A novel T cell receptor single-chain signaling complex mediates antigen-specific T cell activity and tumor control. Cancer Immunol Immunother. 2014;63(11):1163–1176. doi:10.1007/s00262-014-1586-z.
- Legut M, Dolton G, Mian AA, Ottmann OG, Sewell AK. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood. 2018;131(3):311–322. doi:10.1182/blood-2017-05-787598.