
ABSTRACT
ADAM10 and ADAM17 expression and soluble PD-L1 (sPD-L1) predict poor prognosis in many malignancies, including in patients treated with PD-(L)1 inhibitors. The mechanism of soluble PD-L1 production and its effects are unknown. Here we uncover a novel mechanism of ADAM10- and ADAM17-mediated resistance to PD-(L)1 inhibitors. ADAM10 and ADAM17 cleave PD-L1 from the surface of malignant cells and extracellular vesicles. This cleavage produces an active sPD-L1 fragment that induces apoptosis in CD8 + T cells and compromises the killing of tumor cells by CD8 + T cells. Reduced tumor site PD-L1 protein-to-mRNA ratios predict poor outcomes and are correlated with elevated ADAM10 and ADAM17 expression in multiple cancers. These results may explain the discordance between PD-L1 immunohistochemistry and PD-(L)1 inhibitor response. Thus, including ADAM10 and ADAM17 tissue staining may improve therapy selection. Furthermore, treatment with an ADAM10/ADAM17 inhibitor may abrogate PD-(L)1 inhibitor resistance and improve clinical responses to PD-(L)1 immunotherapy.
Introduction
ADAM10 and ADAM17 (TACE) contribute to cancer mortality. High expression of one or both of these closely related metzincin metalloproteinases portends unfavorable outcomes in biliary, breast, cervical, gastric, hepatocellular, lung, nasopharyngeal, ovarian, pancreatic, skin, and urothelial malignancies.Citation1–Citation5 They also predict treatment resistance in breast, colorectal, hepatocellular, and lung cancers.Citation6–Citation9
Long-standing dogma held that metalloproteases contribute to tumor growth and metastasis through enzymatic degradation of the extracellular matrix and tumor cell attachments.Citation10 More recently they have also been shown to alter signaling through modification of surface ligands and receptors such as Notch, HER2, EGFR, and NKG2D.Citation11–Citation13 Metalloprotease inhibitors have shown promise in preventing post-radiation resistance in non-small cell lung cancer and in treating breast cancer.Citation6,Citation9 The effect of ADAM10 and ADAM17 inhibition on antitumor immunity is not well understood.
In this study we explore the role of ADAM10 and ADAM17 at the PD-1/PD-L1 immune checkpoint and in driving resistance to anti-PD-(L)1 therapy. The programmed cell death protein-1 (PD-1) immune checkpoint is a critical regulatory mechanism in antitumor immunity.Citation14 Programmed death-ligand 1 (PD-L1, CD274, B7H1) on tumor cells and other cells engages PD-1 on immune cells to downregulate essential anti-cancer immune functions. Clinical PD-(L)1 inhibitors like pembrolizumab, nivolumab, and atezolizumab obstruct this PD-L1-mediated immunosuppression.Citation15 These monoclonal antibodies against PD-1 or PD-L1 improve survival for patients with melanoma, non-small cell lung cancer, renal cell carcinoma, bladder cancer, colorectal cancer, and other advanced malignancies.Citation16
Despite the successes of PD-(L)1 inhibitors against these cancers, only a fraction of patients benefit from treatment.Citation17 In some indications, tumor PD-L1 staining is performed to select patients who are most likely to respond. Surprisingly, many tumors that express PD-L1 are resistant to PD-(L)1 inhibitors; conversely, some PD-L1-negative tumors respond to PD-1/PD-L1 blockade.Citation18
Understanding mechanisms of PD-(L)1 inhibitor resistance is crucial in improving antitumor immunity and survival. Here, we report a novel anti-PD-(L)1 resistance mechanism mediated by metalloproteases ADAM10 and ADAM17. We show that tumor ADAM10 and ADAM17 cleave the PD-L1 ectodomain from tumor cells and extracellular vesicles (EVs) to form soluble PD-L1 (sPD-L1). sPD-L1 induces apoptosis in human CD8 + T cells, which can be overcome by ADAM10/ADAM17 inhibition.
Materials and methods
Cell culture
Malignant cell lines 786–0, Karpas-299, and Du145 (ATCC) were grown in RPMI 1640 (Invitrogen) supplemented by 10% FBS and maintained at 37°C in 5% CO2 at the concentrations specified. Malignant cell lines A549 (ATCC) and Mel-B7H1Citation14 were grown in DMEM (Gibco) supplemented by 10% FBS and maintained at 37°C in 5% CO2 at the concentrations specified. Cells were used within 4–6 passages after thawing. Mycoplasma testing was performed and negative on each cell line used.
Cell lines were treated as specified with ARP-101, doxycycline hyclate, MMP-9 Inhibitor I, Ro 32–3555, TAPI-0, TAPI-2, UK370106 (Santa Cruz); Aprotinin, E-64, GI254023X, Pepstatin (Sigma); DMSO, PBS (Mediatech); and/or recombinant human IFN-γ (Biosource). Inhibitor concentrations, listed in Supplemental Table 4, were based on common concentrations in the literature. Supernatants were isolated from cell culture by ultracentrifugation. Cell lines were also treated as specified with negative control, ADAM10, or ADAM17 siRNA pools (Dharmacon) with lipofectamine (Fisher) per manufacturer protocols.
Human PBMCs from healthy donors were isolated by Lymphoprep gradient centrifugation and CD8 + T lymphocytes were further purified by negative selection using a magnetic enrichment method (StemCell Technologies). Purified CD8 + T cells (2 × 105 cells/200 µl) were stimulated with anti-CD3 antibodies over 24 h and treated as specified with supernatants, recombinant Fc-PD-L1 (R&D Systems), nivolumab (BMS), pembrolizumab (Merck), and/or atezolizumab (Genentech) at the indicated concentrations. PBMCs were stimulated with PHA-L (Sigma) over 24 h. Viable cells were counted by trypan blue exclusion or analyzed by flow cytometry as indicated.
ELISA
Enzyme-linked immunosorbent assay (ELISA) was performed as previously published.Citation19 In brief, paired mouse IgG2 monoclonal antibody clones H1A and B11 against extracellular human PD-L1 were utilized in a capture-detection plate assay using biotinylation and HRP-streptavidin detection. This assay is specific for sPD-L1 and does not exhibit cross-reactivity to other B7-H homologues. Concentrations were determined by optical density (OD) measurements along a known standard curve of recombinant human PD-L1.
Flow cytometry
Antibodies and reagents used for flow cytometry were Annexin-V-FITC, anti-CD16/32 (BioLegend); anti-PD-L1 (Fisher, clone MIH1); anti-ADAM10 (Abcam, clone 11G2); anti-ADAM17 (Lifespan Biosciences, clone 1F6); anti-human IgG (Biosource); anti-mouse IgG (Invitrogen); isotype controls (eBioScience); TMRE-PE (Abcam); and BD Wash/Perm (Becton Dickenson). Atezolizumab (Genentech) was conjugated with Alexafluor-647 per the manufacturer’s protocol (Invitrogen). Flow cytometry was performed on a CytoFLEX LX flow cytometer (Beckman Coulter) and analyzed using FlowJo software (Treestar).
Flow cytometry of extracellular vesicles (EVs) was performed on isolated supernatants from cell culture. Supernatants were initially centrifuged to remove non-adherent cells. These supernatants were then centrifuged according to existing protocols and analyzed using an A50-Micro Plus Nanoscale Flow Cytometer (Apogee FlowSystems) gating for mid-intensity light angle light scatter (LALS) and marker of interest.Citation20 Nanoscale flow cytometer calibration was performed using a standard reference bead mix.
Immunohistochemistry/immunofluorescence
Tissue samples from consented human subjects were obtained as formalin-fixed paraffin-embedded blocks from clinical archives. This process was overseen and approved by the Mayo Clinic Institutional Review Board (IRB, protocol number 15–000934). For immunohistochemistry (IHC), specimens were sectioned (5 µm) and stained with antibodies against ADAM10 (Abcam, clone 11G2), ADAM17 (Lifespan Biosciences, clone 1F6), or PD-L1 (Cell Signal, clone E1L3 N). Immunohistochemistry slides were counterstained with hematoxylin. Images were reviewed by a pathologist and scored positive for moderate to intense staining. For immunofluorescence (IF), cells were cultured as specified, fixed in paraffin, and counterstained with DAPI and rhodamine-conjugated wheat germ agglutinin (WGA-rhodamine) prior to imaging by confocal microscopy.
Calcein immunofluorescence-based survival was assayed by automated live microscopy at 10-min intervals. In brief, 786–0 cells were treated with control, ADAM10, and/or ADAM17 siRNA. Cells were then washed and loaded with 5uM calcein for 30 min and subsequently washed and treated with PHA-L-activated PBMCs from healthy donors at a 10:1 ratio in 96-well plates. Automated microscopy was performed at 10-min intervals in each of 6 wells per treatment group over 4 h with the EVOS cell imaging system (Thermo Fisher). Calcein-positive densitometry was quantified using ImageJ (NIH) and plotted versus time zero control.
Cancer Genome Atlas data
Level 3 data including sequence per million mapped fragments (FPKM) transcript data from validated RNA-seq experiments and PD-L1 protein levels from validated reverse phase protein array (RPPA) experiments was downloaded for all available patients in The Cancer Genome Atlas (TCGA) database. RPPA data were normalized to scale and a simple PD-L1 protein-to-mRNA ratio for each tumor was calculated by dividing reverse phase protein array (RPPA) PD-L1 protein levels by RNA-seq CD274 levels. Survival analysis was performed by Cox proportional hazards modeling and all data were right censored for analysis. PD-L1 protein-to-mRNA ratio cutoffs were generated by iterative Cox proportional hazards modeling without correction for age or stage of tumor as described.Citation21 Regression modeling was performed for each cancer subset using the base R! linear regression model. P < .05 was considered statistically significant.
Statistical analysis
Statistical consultation was generously provided by Nathan Foster and Paul Novotny of the Mayo Clinic Center for Clinical and Translational Science (CCaTS). Bioinformatics support was provided by the Mayo Clinic Division of Biomedical Statistics and Informatics. All statistical analyses were performed using R! Statistical Software (R Foundation). Unpaired student’s t-test assessed statistical differences in experimental groups except where otherwise indicated. Figures comprising box plots show quartile values and individual data points. Mean values and 95% confidence intervals (CI) are indicated in corresponding supplemental figures and tables. P < .05 was considered statistically significant. In figures, p values are denoted <0.05 with *, <0.01 with **, and <0.001 with ***.
Results
Low PD-L1 protein-to-mRNA ratios predict poor overall survival and are associated with higher metalloprotease mRNA expression in papillary renal cell carcinoma and other malignancies
Disparate tumor PD-L1 protein staining and clinical response to PD-(L)1 inhibitor therapy frustrates regulators, pathologists, and clinicians.Citation18 We hypothesized that post-translational tumor PD-L1 loss may explain both the variability of PD-L1 expression and the discordance between PD-(L)1 staining and clinical responses to PD-(L)1 inhibitors. To determine whether PD-L1 protein levels are commensurate with CD274 (PD-L1) mRNA expression in human tumors, we queried The Cancer Genome Atlas (TCGA) for normalized reverse phase protein array (RPPA) PD-L1 protein levels and for normalized CD274 RNA-seq transcript sequences per million mapped fragments (FPKM) and calculated a PD-L1 protein-to-mRNA ratio for each sample. A violin plot of these ratios by cancer subtype is shown ()). Most cancer subtypes demonstrate substantial variation in PD-L1 protein-to-mRNA ratios.
Figure 1. Low PD-L1 protein-to-mRNA ratios predict poor overall survival and higher metalloprotease mRNA expression in papillary renal cell carcinoma and other malignancies. (a) Cases from The Cancer Genome Atlas (TCGA) public dataset were queried for PD-L1 protein levels by reverse phase protein array (RPPA) and for CD274 (PD-L1) RNA-seq transcript sequence per million mapped fragments (FPKM). A PD-L1 protein-to-mRNA ratio was calculated for each tumor sample. Variability of PD-L1 protein-to-mRNA ratios is shown by cancer subtype in a violin plot. (b) Cases in each cancer subtype were divided into high versus low PD-L1 protein-to-mRNA ratio groups. Survival for each group, controlling for age and tumor stage at diagnosis, was compared by Cox proportional hazards modeling and reported by forest plot (see ). (c) Cases of papillary renal cell carcinoma were divided by high versus low protein-to-mRNA ratio (cutoff 2.56E-6) and survival was compared, controlling for age and stage at diagnosis. (d) ADAM10 and ADAM17 expression (RNASeq normalized FPKM) in each papillary renal cell carcinoma case from TCGA were plotted against PD-L1 protein-to-mRNA ratios and correlation was analyzed by linear regression. ADAM17 expression correlated inversely with PD-L1 protein-to-mRNA ratios (p < .0001). Additional analyses across the TCGA dataset are reported in Supplemental Figure 1 and Supplemental Tables 1–2. Corresponding cancer subtype names and data are listed in . *** P < .001
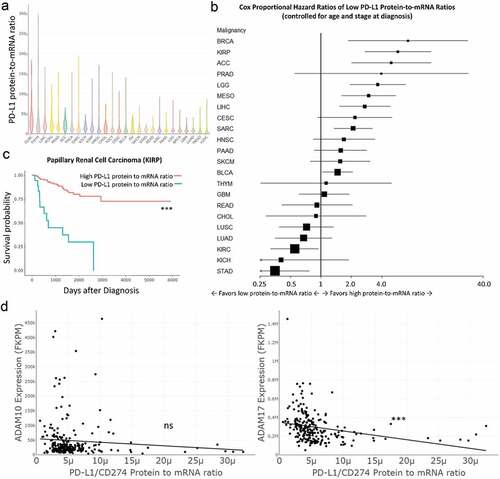
We next considered whether tumor PD-L1 protein-to-mRNA ratio might predict overall survival in these malignancies. We performed Cox proportional hazards testing between groups of high and low protein-to-mRNA ratios for each malignancy, controlling for age and tumor stage at diagnosis. A forest plot and table reporting group size (n), cutoff values, and hazard ratios of death with 95% confidence intervals (CI) are shown (), ). Low PD-L1 protein-to-mRNA ratios predicted poor survival in adrenocortical adenoma, urothelial bladder cancer, breast cancer, papillary renal carcinoma, low grade glioma, hepatocellular carcinoma, and mesothelioma when controlling for age and tumor stage. Low PD-L1 protein-to-mRNA ratios predicted improved outcomes for renal clear cell carcinoma and stomach adenocarcinoma. Survival between groups without regard to age or stage at diagnosis was also analyzed with similar results (Supplemental Figure 1a, Supplemental Table 1).
Table 1. Low PD-L1 protein-to-mRNA ratios predict worse survival in multiple malignancies. Cox proportional hazards modeling of death from tumors expressing high versus low PD-L1 protein-to-mRNA ratios by cancer subtype controlling for age and stage at diagnosis. See Figure 1B
Given these findings, we hypothesized that low PD-L1 protein levels in cells expressing PD-L1 mRNA may result from surface PD-L1 ectodomain shedding to produce soluble PD-L1. We previously showed that renal carcinoma cells produce a soluble form of PD-L1 (sPD-L1) and that patients with high serum sPD-L1 experience worse outcomes in renal cancer.Citation19 In our Cox proportional hazards models, low PD-L1 protein-to-mRNA ratios predict strikingly poor outcomes in papillary renal cell carcinoma ()).
The source of soluble PD-L1 is uncertain. While others have reported secreted PD-L1 splice variants,Citation22 we hypothesized that PD-L1 may be cleaved from the surface of tumor cells. In particular, closely related metzincin metalloproteinases ADAM10 and ADAM17 commonly cleave immune cell surface ligands. To test the hypothesis that ADAM10 and ADAM17 cleave surface PD-L1, we compared normalized RNASeq FPKM for ADAM10 and ADAM17 in tumor samples with PD-L1 protein-to-mRNA ratios in papillary renal cell carcinoma by linear regression model ()). ADAM17 mRNA expression levels showed significant inverse correlation with PD-L1 protein-to-mRNA ratios in papillary kidney cancer. Similar analyses were performed on all other TCGA cancer subsets (Supplemental Figure 1b, Supplemental Table 2). In all 26 cancer subtypes in the dataset, ADAM10 and/or ADAM17 transcripts were significantly inversely correlated with tumor sample PD-L1 protein-to-mRNA ratios. While correlative in nature and encompassing whole tumor beds including stromal cells, these results suggested a possible role for ADAM10 and/or ADAM17 in PD-L1 removal from the tumor cell surface.
ADAM10 and ADAM17 (TACE) cleave sPD-L1 from the surface of tumor cells
To test our hypothesis that tumor surface proteases ADAM10 and ADAM17 cleave surface PD-L1 to release sPD-L1, we screened multiple malignant cell lines with a variety of related metalloprotease inhibitors (including aderbasib, TAPI-0, TAPI-2, and GI-254023X) and with broad protease inhibitors (Supplemental Figure 2). In these cell lines, we found that inhibitors of metalloproteases ADAM10 and ADAM17 consistently and significantly reduce tumor cell line supernatant sPD-L1 at 48 h.
Focusing on our renal cancer model, we treated wild-type 786–0 renal cancer cells with DMSO vehicle control versus protease inhibitors over 48 h and assayed cell supernatants for sPD-L1 by ELISA. 786–0 renal carcinoma cells produced copious sPD-L1 in vitro at high cell titers and on interferon-γ treatment, but an inhibitor of ADAM10 and ADAM17 reduced measurable supernatant sPD-L1 (), Supplemental Table 3). Inhibiting ADAM10 and ADAM17 did not alter PD-L1 mRNA levels (Supplemental Figure 3). 786–0 renal cancer cell line sPD-L1 production varied predictably with incubation time, cell concentration, and inhibitor concentration (Supplemental Figure 4a-b). 786–0 and other cell lines that produce sPD-L1 express surface ADAM10 and ADAM17 by flow cytometry (Supplemental Figure 4c).
Figure 2. ADAM10 and ADAM17 (TACE) cleave sPD-L1 from the surface of tumor cells. (a) Wild-type 786–0 renal cancer cells were grown in conditions known to induce sPD-L1 production (density 106 cells/ml with interferon-γ or density 107 cells/ml with vehicle control) in the absence or presence of ADAM10/ADAM17 inhibitor TAPI-0 over 48 h. Supernatants were collected and assayed for sPD-L1 by ELISA. Box plots shown. Additional cell lines, inhibitors, and concentrations shown in Supplemental and . (b) Schematic for panels C-D. 786–0 cells expressing C-terminus PD-L1 GFP (786-0-PDL1-GFP) at 3 · 104 cells/ml were labeled with Alexafluor-647-conjugated atezolizumab (ATZ-Alexa647) and then treated as indicated prior to assay. (c) ATZ-Alexa647-labeled 786-0-PDL1-GFP cells treated with DMSO vehicle or TAPI-2 were assayed for maintained surface PD-L1 (GFPhigh/Alexa647+) by flow cytometry at 24 h. ATZ-Alexa647-labeled 786-0-PDL1-GFP cells treated with DMSO vehicle versus TAPI-2 were visualized for nuclei (DAPI, blue), C-terminus PD-L1 (PD-L1-GFP, green), lipid membranes (WGA-rhodamine, red), and N-terminus PD-L1 (Atezolizumab-A647, purple) by immunofluorescence at 1 h and 24 h. Vehicle control-treated cells showed perinuclear localization of residual GFP (gray arrows). TAPI-2 treated cells showed increased total PD-L1 and starting surface PD-L1 (white arrows). Box plots quantifying flow cytometry shown, compared statistically by Student’s t-test. (d) ATZ-Alexa647-labeled 786-0-PDL1-GFP cells treated with control siRNA versus ADAM10/ADAM17 siRNA versus TAPI-2 were visualized for nuclei (DAPI, blue), C-terminus PD-L1 (PD-L1-GFP, green), lipid membranes (WGA-rhodamine, red), and N-terminus PD-L1 (Atezolizumab-A647, purple) by immunofluorescence at 24 h. Corresponding siRNA and flow cytometry data in Supplemental Figure 5A-D. Box plots quantifying flow cytometry shown, compared statistically by Student’s t-test. (e) Karpas-299 cells were treated with vehicle control versus TAPI-2, recombinant human ADAM10, or recombinant human ADAM17 over 24 h and PD-L1 positive cells were measured by flow cytometry. Box plots quantifying flow cytometry mean fluorescence intensity (MFI) shown, compared statistically by Student’s t test. (f) A549 lung carcinoma cells were treated with vehicle control versus recombinant human ADAM17 (TACE) over 24 h and sPD-L1 was measured from cell supernatants by ELISA. Box plots and statistical data including p value versus control, mean, and 95% confidence intervals by unpaired Student’s t-test are shown. *** P < .001, ** P < .01, * P < .05. Additional-related experimental data are in Supplemental Tables 3–4, Supplemental Figures 5–6
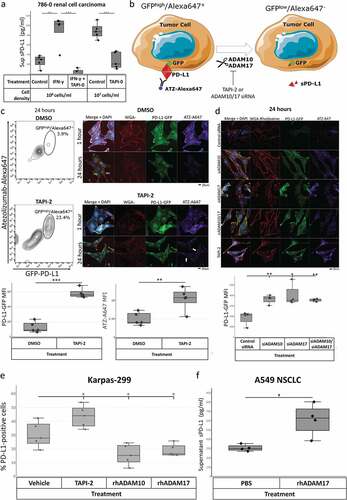
786-0-PD-L1-GFP renal carcinoma cells carry a PD-L1 overexpression construct with an intracellular C-terminus GFP tag. To further characterize PD-L1 cleavage by ADAM10 and ADAM17, we labeled N-terminus surface PD-L1 on 786-0-PD-L1-GFP cells with Alexafluor-647-conjugated anti-PD-L1 antibody atezolizumab (ATZ-Alexa647) at time 0 and treated with DMSO vehicle control versus metalloprotease inhibitor TAPI-2 ()). After 24 h, we measured fluorescence by flow cytometry and confocal microscopy. By flow cytometry, 786-0-PD-L1-GFP cells treated with this broad inhibitor maintained more intact PD-L1 as evidenced by abundant GFPhigh/Alexa647+ cells versus vehicle-treated control (), Supplemental Figure 5a-b). 786-0-PD-L1-GFP cells visualized by confocal microscopy at one-hour post-treatment maintained both total cell PD-L1 (GFP-PD-L1, green) and starting surface PD-L1 (ATZ-A647, purple) which were localized to the cell surface (WGA-rhodamine, red) by confocal microscopy ()). At 24 h, vehicle control-treated cells lost both starting surface PD-L1 and total PD-L1 with perinuclear localization of residual GFP. In contrast, TAPI-2-treated cells at 24 h maintained both total PD-L1 and starting surface PD-L1. Similar results were seen by immunofluorescence and flow cytometry when ADAM10 and ADAM17 were knocked down with pooled siRNA (), Supplemental Figure 5 c-d). Similar results were seen in the MB-MDA-231 breast cancer cell line (Supplemental Figure 6a). Inhibition with ADAM10-specific peptide TIMP-1 and ADAM17-specific function-blocking monoclonal antibody D1A similarly maintained 786-0-PD-L1-GFP surface PD-L1 at 24 h versus vehicle control and prevented sPD-L1 generation (Supplemental Figure 6b).
Karpas-299 cells also release ADAM10-cleaved sPD-L1 (Supplemental Figure 3b, 4c). We treated these cells with vehicle control, TAPI-2, or exogenous recombinant ADAM10 or ADAM17 and measured surface PD-L1 staining by flow cytometry ()). TAPI-2-mediated ADAM10/ADAM17 inhibition increased surface PD-L1 staining, while recombinant ADAM10 and ADAM17 each significantly reduced surface PD-L1 staining. Unlike 786–0 renal carcinoma cells, Karpas-299 cells, and other cell lines tested, A549 lung carcinoma cells express surface PD-L1 but neither ADAM10 nor ADAM17 (Supplemental Figure 4c). A549 cells do not produce high levels of sPD-L1. We treated these cells with exogenous recombinant ADAM17 in culture medium over 48 h. Exogenous ADAM17 induced A549 sPD-L1 shedding as measured by ELISA ()). Notably, PD-L1 protein-to-mRNA ratio did not correlate with survival in patients with lung cancer in The Cancer Genome Atlas dataset (Supplemental Figure 1b).
Primary melanoma ADAM10 and ADAM17 staining correlates negatively with PD-L1 staining
ADAM10/ADAM17-mediated tumor sPD-L1 cleavage occurred in a variety of tumor cell lines as described above. In primary tumors, cell PD-L1 staining is often performed on tumor samples prior to treatment. However, tumor PD-L1 staining does not always predict treatment response.Citation18 Furthermore, PD-L1 expression varies within the same tumor both spatially and temporally.Citation23–Citation25
Advanced melanomas are commonly treated with a PD-1 inhibitor such as pembrolizumab for first-line therapy, although most tumors exhibit or acquire resistance to treatment.Citation26 In our TCGA analysis, patients with melanomas demonstrating low PD-L1 protein-to-mRNA ratios trended toward worse outcomes than patients whose tumors featured high PD-L1 protein-to-mRNA ratios ()).
Figure 3. Primary melanoma ADAM10 and ADAM17 staining correlates negatively with PD-L1 staining. (a) Cases of melanoma were divided by high versus low protein-to-mRNA ratio (cutoff 3.59E-6) and overall survival, ADAM10 expression, and ADAM17 expression were compared by Cox proportional hazards testing and unpaired Student’s t-test, respectively. Similar analyses across the TCGA dataset are reported in , Supplemental , and Supplemental Tables 1–2. Primary melanoma samples were stained for ADAM10 (pink, column 1), ADAM17 (pink, column 2), and PD-L1 (brown, column 3) by immunohistochemistry (IHC). 10x and 60x magnification shown for each sample. Dashed rectangle in the 10x magnification images (row 1) denotes the area of 60X magnification (row 2). An asterisk (*) denotes moderate to intense staining as scored by a pathologist. (b) In Case 1, melanoma cells stained positively for PD-L1 but not for ADAM10 or ADAM17. (c) In Case 2, melanoma cells stained positively for ADAM10 but not for ADAM17 or PD-L1. Additional cases in Supplemental Figure 7A-D. Fisher’s exact test showed significant negative correlation between ADAM10/ADAM17 and PD-L1 staining (odds ratio 0, CI 0–0.832, p = .018; see Supplemental Figure 7E)
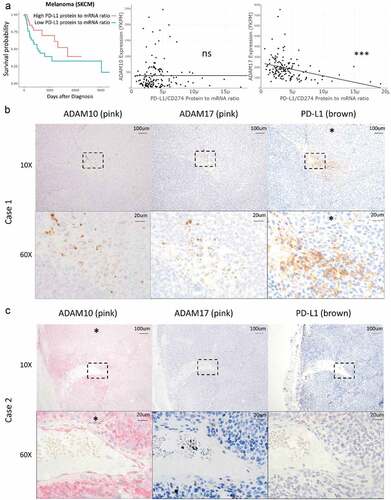
To assess whether ADAM10 or ADAM17 activity contributes to tumor surface PD-L1 staining variability, we stained sequential melanoma sections with antibodies against ADAM10, ADAM17, and PD-L1 (-c), Supplemental Figure 7). In the eight samples stained from different patients with melanoma, PD-L1 and ADAM10 or ADAM17 staining were mutually exclusive. Samples positive for PD-L1 were negative for ADAM10 and ADAM17 (), Supplemental Figure 7A). Conversely, samples positive for ADAM10 and/or ADAM17 were negative for PD-L1 (), Supplemental Figure 7B-D). This negative correlation was statistically significant by Fisher’s exact test (Supplemental Figure 7E). As a positive internal control, ADAM10 was observed in endothelial cells surrounding blood vessels.
Tumor-derived sPDL1 induces activated human CD8 + T cell apoptosis and competes with PD-(L)1 inhibitors for effect on CD8 + T cells
We hypothesized that ADAM10 and ADAM17-generated tumor-derived sPD-L1 may induce human CD8 + T cell death. To characterize sPD-L1-mediated immune cell death, we treated CD8 + T cells from healthy human donors over 48 h with recombinant Fc fusion PD-L1 protein (), Supplemental Table 5). Recombinant Fc-PD-L1 significantly reduced CD8 + T cell survival.
Figure 4. Tumor-derived sPD-L1 induces activated human CD8 + T cell death and competes with PD-(l)1 inhibitors for effect on CD8 + T cells. Human CD8 + T cells from unique healthy donors were cultured (a) in the presence of PBS control versus 5 µg/ml or 10 µg/ml recombinant sPD-L1 or (b) in the presence of sPD-L1-rich Karpas-299 cell line supernatant with or without PD-(L)1 inhibitors. Cell survival was measured by trypan blue staining. CD8+ human T cells from unique healthy donors were cultured in the presence of supernatant from wild type versus PD-L1 knockout 786–0 cells in the presence of varying concentrations of PD-1 inhibitor pembrolizumab. Apoptosis was measured by TMRE and Annexin-V staining on flow cytometry (c), percent TMRE-negative Annexin-V-positive apoptotic cells in treatment groups are reported (d). Additional flow diagrams in Supplemental Figure 8. A model of ADAM10/ADAM17-induced sPD-L1-mediated PD-(L)1 resistance is diagrammed (e). Healthy human PBMCs from unique donors were treated with supernatant from wild type versus PD-L1-knockout 786–0 cells over 4 h. PBMCs were then isolated and added to calcein-labeled 786–0 PD-L1 knockout cells at a 10:1 ratio and cell survival was visualized by microscopy and quantified at 0, 1, and 2 h (f-g). Cells in this figure were numbered where helpful. Statistical analyses performed with unpaired Student’s t-test (see Supplemental Table 5). *** P < .001, ** P < .01, * P < .05
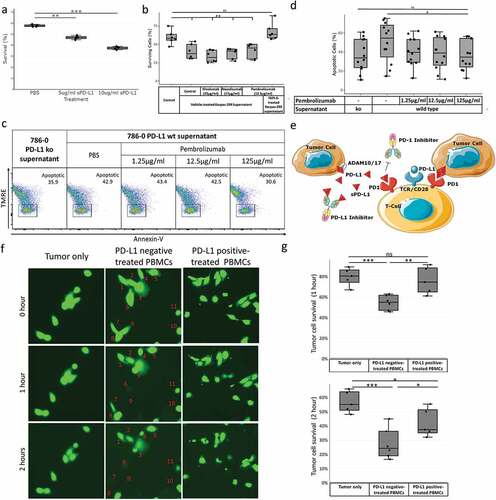
To determine whether ADAM10- and ADAM17-generated sPD-L1 induce immune cell death despite treatment with PD-(L)1 inhibitors, we treated CD8 + T cells from healthy human donors with sPD-L1-rich supernatant from Karpas-299 cells for 48 h with or without PD-(L)1 inhibitors and measured cell survival by trypan blue staining ()). sPD-L1-rich supernatants reduced CD8 + T cell survival despite pre-treatment with PD-(L)1 inhibitors. Conversely, Karpas-299 cell supernatant generated in the presence of inhibitor TAPI-2 to prevent sPD-L1 production did not induce CD8 + T cell death.
We further hypothesized that high doses of PD-1 inhibitor may prevent sPD-L1-mediated CD8 + T cell death. We treated CD8+ human T cells isolated from 12 different healthy human donors with increasing concentrations of PD-1 inhibitor pembrolizumab in the presence of supernatant from 786 to 0 wild type versus 786-0-PD-L1-null cells and measured Annexin-V-positive, TMRE-negative apoptotic cells by flow cytometry (-d), Supplemental Figure 8). High doses of pembrolizumab reduced CD8 + T cell apoptosis in each matched donor. In a confirmatory experiment, the PD-L1 inhibitor atezolizumab at high concentrations also rescued CD8 + T cells from sPD-L1-mediated apoptosis (Supplemental Figure 9). Spiking supernatants with inhibitor did not reduce CD8 + T cell apoptosis. These results suggest that ADAM10 and ADAM17 cleave surface PD-L1 to produce sPD-L1, which in turn induces CD8 + T cell apoptosis and prevents PD-(L)1 inhibitor function ()).
We next sought to determine whether sPD-L1 exposure broadly dampens antitumor immunity. We isolated peripheral blood mononuclear cells (PBMCs) and pre-treated them with supernatant from wild-type versus PD-L1-deficient786-0 cells over 4 h. We also loaded 786–0 PD-L1-deficient cells with calcein dye. These calcein-loaded PD-L1-null tumor cells were then washed and treated with the above PBMCs at 10:1 versus medium control and visualized by microscopy for tumor calcein positivity at 0, 60, and 120 min ()). PD-L1-negative supernatant-treated PBMCs significantly reduced tumor cell survival at 60 and 120 min versus media control and PD-L1 positive supernatant-treated PBMCs.
We further considered whether ADAM10 and/or ADAM17 activity in the tumor itself prevents tumor cell killing by peripheral blood mononuclear cells (PBMCs). We knocked down ADAM10 and/or ADAM17 expression in 786–0 wild-type and PD-L1-null renal cancer cells using siRNA, then washed and loaded with calcein dye. We cocultured these tumor cells with healthy peripheral blood mononuclear cells (PBMCs) at a 10:1 ratio and measured the loss of calcein-positive cells by automated fluorescence microscopy at 10-min intervals (Supplemental Figure 10). Knockdown of tumor cell ADAM10, ADAM17, or both significantly increased PBMC-mediated tumor cell death in wild-type, but not PD-L1-deficient, 786–0 cells. This suggests that this resistance is mediated by ADAM10 and/or ADAM17 and requires PD-L1.
Tumor cells may secrete PD-L1-positive extracellular vesicles (EVs) to downregulate tumor immunity.Citation27 Both ADAM10 and ADAM17 are known extracellular vesicle components.Citation28 To investigate whether ADAM10 or ADAM17 cleave PD-L1 from the surface of EVs, we labeled 786–0 PD-L1-overexpressing cells with Alexafluor-647-conjugated atezolizumab and incubated over 24 h. We then isolated EVs from the cell supernatant and incubated an additional 24 h in the presence of vehicle control versus TAPI-2 and analyzed EVs by nanoflow cytometry (Supplemental Figure 11). Treatment of vesicles with TAPI-2 significantly increased the number of ATZ-positive EVs remaining after 24 h, suggesting that ADAM10 and ADAM17 continue to cleave PD-L1 after tumor EV release.
Discussion
In the present work, we showed that tumor-associated metzincin metalloproteinases ADAM10 and ADAM17 cleave PD-L1 from the surface of tumor cells to induce apoptosis in CD8 + T cells and dampen anti-tumor immunity. These findings represent a novel mechanism of resistance to PD-(L)1 inhibitors. This newfound activity of tumor ADAM10 and ADAM17 may explain several phenomena observed in human cancers. First, high serum sPD-L1 is a predictor of poor response to PD-1 inhibition in some malignancies and predicts poor outcomes overall in hepatocellular carcinoma, cholangiocarcinoma, lung cancer, melanoma, myeloma, NK T cell lymphoma, and others.Citation29–Citation37 Tumor ADAM10 and ADAM17 cleave sPD-L1 that may mediate this PD-1 inhibitor resistance by inducing CD8 + T cell death. sPD-L1 may also act as a sink for circulating PD-L1 inhibitors (see )).
Second, high sPD-L1 is a positive prognostic indicator in a minority of tumor types, including stomach adenocarcinoma.Citation38,Citation39 Stomach adenocarcinoma is one of the only two malignancies for which our analysis found an association between lower PD-L1 protein-to-mRNA ratios and improved outcomes. Different sPD-L1 cleavage products generated by ADAM10 and/or ADAM17 either alone or in sequence may have different effects on the PD-1 receptor, and different resistance mechanisms may further account for these differences. Future studies of products of proteolytic PD-L1 cleavage – including whether ADAM10 and ADAM17 cleave PD-L1 at the same sites and whether all products exhibit similar activities – are warranted.
Third, some tumors that do not have detectable PD-L1 expression on pathology samples respond to PD-1/PD-L1 inhibition.Citation18 The activity of ADAM10 and ADAM17 may explain negative PD-L1 staining in these cases. It may also explain the spatial and temporal variability of surface PD-L1 expression in some tumors.Citation23–Citation25 Further, sPD-L1 derived from a subset of tumor cells may protect distant malignant cells from anti-tumor immunity. Combined ADAM10/ADAM17/PD-L1 immunohistochemistry and serum sPD-L1 may thus better select patients for PD-(L)1 inhibitor therapy in future studies.
While these data suggest that tumor-derived soluble PD-L1 may play a role in tumor immunosuppression, there is currently no animal model of sPD-L1-induced resistance. It is also unknown how these findings relate to previously published secreted PD-L1 splice variants.Citation22 While we observed that PD-L1 is also shed from the surface of extracellular vesicles, further work is required to determine the relative contribution of exosome-derived sPD-L1 to resistance. It is also uncertain whether removal of either ADAM10 or ADAM17 alone, as in our siRNA studies but not in our peptide and antibody inhibitor experiments, is sufficient to rescue PD-(L)1 inhibitor sensitivity. This may reflect the difference between RNA-level silencing of enzyme expression versus cell surface-limited competitive inhibition of ADAM10 and ADAM17 by peptide and antibody in these experiments. Given differences in tumor subtype expression and the many modes of regulation of ADAM10 and ADAM17 activity, different tumors may respond differently to blockade of both or either of these enzymes. Clinical studies are warranted to validate these findings in patients.
Our findings further support that pre-treatment tumor expression of PD-L1 by immunohistochemistry is insufficient to determine whether a patient may benefit from a PD-(L)1 inhibitor. Combination PD-L1 and ADAM10/ADAM17 immunohistochemistry as well as serum sPD-L1 measurement may further improve therapeutic decision-making. Our results additionally suggest potential novel therapeutic approaches to PD-(L)1 inhibitor-resistant tumors. For instance, patients with selected malignancies could be treated initially with higher doses of a PD-L1 inhibitor that recognizes sPD-L1, although these doses in vivo may be prohibitively high. It is also unknown whether clinical PD-L1 inhibitors bind sPD-L1 fragments equally. Clinical trials would be appropriate to address these questions. Alternatively, selected patients could be treated first with plasma exchange to remove circulating sPD-L1 followed by a PD-(L)1 inhibitor. Selected malignancies could otherwise be treated at the source with an inhibitor of ADAM10/ADAM17 in conjunction with a PD-(L)1 inhibitor to improve response. While genetic loss of these proteases is lethal in mice and causes severe inflammatory disease in humans, clinical inhibitors of ADAM10 and ADAM17 have been studied and found to be safe in other indications.Citation40
Authors’ contribution
Jacob Orme developed hypotheses, performed experiments, performed data analysis and programming, and wrote the article. Khalid Jazieh, Tiancheng Xie, Susan Harrington, Benjamin Madden, Cristine Charlesworth, and Fabrice Lucien performed experiments. Matthew Ball read pathology. Xin Liu and Yanli Li gave experimental materials and laboratory support. Bharath Wootla provided technical input and illustrations. Tariq Azam organized melanoma cases. Aaron Mansfield, Roxana Dronca, and Haidong Dong contributed to discussion of the concepts, providing funding, mentoring experimental designs, revising the manuscript, and pathology samples. J.C. Villasboas reviewed and provided input in the manuscript and provided mentoring for the project.
Disclosure of potential conflicts of interest
Intellectual property has been filed addressing discoveries disclosed in this manuscript. ASM notes honoraria to Mayo Clinic for advisory boards from Abbvie, BMS, Genentech. The authors report no other relevant conflicts of interest.
Supplemental Material
Download ()Acknowledgments
Statistical guidance was provided generously by Nathan Foster and Paul Novotny of the Mayo Clinic Center for Clinical and Translational Science (CCaTS). Technical guidance for working with TCGA data was provided generously by Jeran Stratford. The authors acknowledge the help of Jacob Hirdler in performing an experiment. Some illustrations were created using Servier Medical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License; https://smart.servier.com. The results shown here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357(6352):eaan2507. doi:10.1126/science.aan2507.
- Buchanan PC, Boylan KLM, Walcheck B, Heinze R, Geller MA, Argenta PA, Skubitz APN. Ectodomain shedding of the cell adhesion molecule nectin-4 in ovarian cancer is mediated by ADAM10 and ADAM17. J Biol Chem. 2017;292:6339–11. doi:10.1074/jbc.M116.746859.
- Ni -S-S, Zhang J, Zhao W-L, Dong X-C, Wang J-L. ADAM17 is overexpressed in non-small cell lung cancer and its expression correlates with poor patient survival. Tumor Biol. 2013;34:1813–1818. doi:10.1007/s13277-013-0721-3.
- Wang -Y-Y, Ye Z-Y, Li L, Zhao Z-S, Shao Q-S, Tao H-Q. ADAM 10 is associated with gastric cancer progression and prognosis of patients. J Surg Oncol. 2011;103:116–123. doi:10.1002/jso.21781.
- You B, Shan Y, Shi S, Li X, You Y. Effects of ADAM10 upregulation on progression, migration, and prognosis of nasopharyngeal carcinoma. Cancer Sci. 2015;106:1506–1514. doi:10.1111/cas.12800.
- Feldinger K, Generali D, Kramer-Marek G, Gijsen M, Ng TB, Wong JH, Strina C, Cappelletti M, Andreis D, Li J-L, et al. ADAM10 mediates trastuzumab resistance and is correlated with survival in HER2 positive breast cancer. Oncotarget. 2014;5:6633–6646. doi:10.18632/oncotarget.v5i16.
- Van Schaeybroeck S, Kalimutho M, Dunne P, Carson R, Allen W, Jithesh P, Redmond K, Sasazuki T, Shirasawa S, Blayney J, et al. ADAM17-dependent c-MET-STAT3 signaling mediates resistance to MEK inhibitors in KRAS mutant colorectal cancer. Cell Rep. 2014;7:1940–1955. doi:10.1016/j.celrep.2014.05.032.
- Yang C, Jiang F, Xu F, Jiang G. ADAM10 overexpression confers resistance to doxorubicin-induced apoptosis in hepatocellular carcinoma. Tumor Biol. 2012;33:1535–1541. doi:10.1007/s13277-012-0405-4.
- Sharma A, Bender S, Zimmermann M, Riesterer O, Broggini-Tenzer A, Pruschy MN. Secretome signature identifies ADAM17 as novel target for radiosensitization of non-small cell lung cancer. Clin. Cancer Res. 2016;22:4428–4439. doi:10.1158/1078-0432.CCR-15-2449.
- Reiss K, Ludwig A, Saftig P. Breaking up the tie: disintegrin-like metalloproteinases as regulators of cell migration in inflammation and invasion. Pharmacol Ther. 2006;111:985–1006. doi:10.1016/j.pharmthera.2006.02.009.
- Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, Israël A, et al. A novel proteolytic cleavage involved in notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell. 2000;5:207–216. doi:10.1016/S1097-2765(00)80417-7.
- Sahin U, Blobel CP. Ectodomain shedding of the EGF-receptor ligand epigen is mediated by ADAM17. FEBS Lett. 2007;581:41–44. doi:10.1016/j.febslet.2006.11.074.
- Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, Stevanovic S, Rammensee H-G, Steinle A. Tumor-associated MICA is shed by ADAM proteases. Cancer Res. 2008;68:6368–6376. doi:10.1158/0008-5472.CAN-07-6768.
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med. 2002;8:793. doi:10.1038/nm730.
- Dronca RS, Liu X, Harrington SM, Chen L, Cao S, Kottschade LA, McWilliams RR, Block MS, Nevala WK, Thompson MA, et al. T cell bim levels reflect responses to anti–PD-1 cancer therapy. JCI Insight. 2016;1. doi:10.1172/jci.insight.86014.
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi:10.1056/NEJMoa1200690.
- O’Donnell JS, Long GV, Scolyer RA, Teng MWL, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. 2017;52:71–81. doi:10.1016/j.ctrv.2016.11.007.
- Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14:847–856. doi:10.1158/1535-7163.MCT-14-0983.
- Frigola X, Inman BA, Lohse CM, Krco CJ, Cheville JC, Thompson RH, Leibovich B, Blute ML, Dong H, Kwon ED, et al. Identification of a soluble form of B7-H1 that retains immunosuppressive activity and is associated with aggressive renal cell carcinoma. Clin Cancer Res. Clin Cancer Res. April. 2011;1:1915–1923. doi:10.1158/1078-0432.CCR-10-0250.
- Gomes J, Lucien F, Cooper T, Kim Y, Williams K, Liao X, Kaufman L, Lagugné-Labarthet F, Kenyon O, Boysen J, et al. Analytical considerations in nanoscale flow cytometry of extracellular vesicles to achieve data linearity. Thromb Haemost. 2018;118:1612–1624. doi:10.1055/s-0038-1668544.
- Qu P, Crowley J. Fit a grey-zone model with survival data. 2015.
- Mahoney KM, Shukla SA, Patsoukis N, Chaudhri A, Browne EP, Arazi A, Eisenhaure TM, Pendergraft WF, Hua P, Pham HC, et al. A secreted PD-L1 splice variant that covalently dimerizes and mediates immunosuppression. Cancer Immunol Immunother. 2018;1–12. doi:10.1007/s00262-018-2282-1.
- Ilie M, Long-Mira E, Bence C, Butori C, Lassalle S, Bouhlel L, Fazzalari L, Zahaf K, Lalvée S, Washetine K, et al. Comparative study of the PD-L1 status between surgically resected specimens and matched biopsies of NSCLC patients reveal major discordances: a potential issue for anti-PD-L1 therapeutic strategies. Ann. Oncol. 2016;27:147–153. doi:10.1093/annonc/mdv489.
- Mansfield AS, Murphy SJ, Peikert T, Yi ES, Vasmatzis G, Wigle DA, Aubry MC. Heterogeneity of programmed cell death ligand 1 expression in multifocal lung cancer. Clin Cancer Res. 2016;22:2177–2182. doi:10.1158/1078-0432.CCR-15-2246.
- Mansfield A, Dong H. Implications of programmed cell death 1 ligand 1 heterogeneity in the selection of patients with non-small cell lung cancer to receive immunotherapy. Clin Pharmacol Ther. 2016;100:220–222. doi:10.1002/cpt.360.
- Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, Hodi FS, Schachter J, Pavlick AC, Lewis KD, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908–918. doi:10.1016/S1470-2045(15)00083-2.
- Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, Yu Z, Yang J, Wang B, Sun H, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382–386. doi:10.1038/s41586-018-0392-8.
- Stoeck A, Keller S, Riedle S, Sanderson M, Runz S, Le Naour F, Gutwein P, Ludwig A, Rubinstein E, Altevogt P, et al. A role for exosomes in the constitutive and stimulus-induced ectodomain cleavage of L1 and CD44. Biochem. J. 2006;393:609–618. doi:10.1042/BJ20051013.
- Finkelmeier F, Canli O, Tal A, Pleli T, Trojan J, Schmidt M, Piiper A, Kronenberger B, Zeuzem S, Greten FR, et al. High levels of the soluble programmed death-ligand (sPD-L1) identify hepatocellular carcinoma patients with a poor prognosis. Z Gastroenterol. 2015;53:A4_19. doi:10.1055/s-00000094.
- Ha H, Nam A-R, Bang J-H, Park J-E, Kim T-Y, Lee K-H, Han S-W, Im S-A, Kim T-Y, Bang Y-J, et al. Soluble programmed death-ligand 1 (sPDL1) and neutrophil-to-lymphocyte ratio (NLR) predicts survival in advanced biliary tract cancer patients treated with palliative chemotherapy. Oncotarget. 2016;7:76604–76612. doi:10.18632/oncotarget.v7i47.
- Okuma Y, Hosomi Y, Nakahara Y, Watanabe K, Sagawa Y, Homma S. High plasma levels of soluble programmed cell death ligand 1 are prognostic for reduced survival in advanced lung cancer. Lung Cancer. 2017;104:1–6. doi:10.1016/j.lungcan.2016.11.023.
- Zhang J, Gao J, Li Y, Nie J, Dai L, Hu W, Chen X, Han J, Ma X, Tian G, et al. Circulating PD-L1 in NSCLC patients and the correlation between the level of PD-L1 expression and the clinical characteristics. Thorac Cancer. 2015;6:534–538. doi:10.1111/tca.2015.6.issue-4.
- Costantini A, Julie C, Dumenil C, Hélias-Rodzewicz Z, Tisserand J, Dumoulin J, Giraud V, Labrune S, Chinet T, Emile J-F, et al. Predictive role of plasmatic biomarkers in advanced non-small cell lung cancer treated by nivolumab. Oncoimmunology. 2018;e1452581. doi:10.1080/2162402X.2018.1452581.
- Dronca R, Leontovich A, Harrington S, Jegapragasan M, Kottschade L, Nevala W, Enninga E, Markovic S, Dong H. Soluble PD-L1 (sPD-L1) is associated with decreased survival in metastatic melanoma. Pigment Cell Melanoma Res. 2015;28:768–769.
- Dronca RS, Mansfield AS, Liu X, Harrington S, Enninga EA, Kottschade LA, Koo CW, McWilliams RR, Block MS, Nevala WK, et al. Bim and soluble PD-L1 (sPD-L1) as predictive biomarkers of response to anti-PD-1 therapy in patients with melanoma and lung carcinoma. J. Clin. Oncol. 2017;35:11534. doi:10.1200/JCO.2017.35.15_suppl.11534.
- Wang L, Wang H, Chen H, Wang W-D, Chen X-Q, Geng Q-R, Xia Z-J, Lu Y. Serum levels of soluble programmed death ligand 1 predict treatment response and progression free survival in multiple myeloma. Oncotarget. 2015;6:41228–41236. doi:10.18632/oncotarget.v6i38.
- Wang H, Wang L, Liu W-J, Xia Z-J, Huang H-Q, Jiang W-Q, Li Z-M, Lu Y. High post-treatment serum levels of soluble programmed cell death ligand 1 predict early relapse and poor prognosis in extranodal NK/T cell lymphoma patients. Oncotarget. 2016;7:33035–33045. doi:10.18632/oncotarget.8847.
- Sideras K, de Man RA, Harrington SM, Polak WG, Zhou G, Schutz HM, Pedroza-Gonzalez A, Biermann K, Mancham S, Hansen BE, et al. Circulating levels of PD-L1 and galectin-9 are associated with patient survival in surgically treated hepatocellular carcinoma independent of their intra-tumoral expression levels. Sci Rep. 2019;9:10677. doi:10.1038/s41598-019-47235-z.
- Zheng Z, Bu Z, Liu X, Zhang L, Li Z, Wu A, Wu X, Cheng X, Xing X, Du H, et al. Level of circulating PD-L1 expression in patients with advanced gastric cancer and its clinical implications. Chin J Cancer Res. 2014;26:104–111. doi:10.3978/j..1000-9604.2014.02.08.
- Blaydon DC, Biancheri P, Di W-L, Plagnol V, Cabral RM, Brooke MA, van Heel DA, Ruschendorf F, Toynbee M, Walne A, et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N Engl J Med. 2011;365:1502–1508. doi:10.1056/NEJMoa1100721.