
ABSTRACT
Recent advances in novel immune strategies, particularly chimeric antigen receptor (CAR)-bearing T-cells, have shown limited efficacy against glioblastoma (GBM) in clinical trials. We currently have an incomplete understanding of how these emerging therapies integrate with the current standard of care, specifically radiation therapy (RT). Additionally, there is an insufficient number of preclinical studies monitoring these therapies with high spatiotemporal resolution. To address these limitations, we report the first longitudinal fluorescence-based intravital microscopy imaging of CAR T-cells within an orthotopic GBM preclinical model to illustrate the necessity of RT for complete therapeutic response. Additionally, we detail the first usage of murine-derived CAR T-cells targeting the disialoganglioside GD2 in an immunocompetent tumor model. Cell culture assays demonstrated substantial GD2 CAR T-cell-mediated killing of murine GBM cell lines SB28 and GL26 induced to overexpress GD2. Complete antitumor response in advanced syngeneic orthotopic models of GBM was achieved only when a single intravenous dose of GD2 CAR T-cells was following either sub-lethal whole-body irradiation or focal RT. Intravital microscopy imaging successfully visualized CAR T-cell homing and T-cell mediated apoptosis of tumor cells in real-time within the tumor stroma. Findings indicate that RT allows for rapid CAR T-cell extravasation from the vasculature and expansion within the tumor microenvironment, leading to a more robust and lasting immunologic response. These exciting results highlight potential opportunities to improve intravenous adoptive T-cell administration in the treatment of GBM through concurrent RT. Additionally, they emphasize the need for advancements in immunotherapeutic homing to and extravasation through the tumor microenvironment.
Introduction
Glioblastoma (GBM) is the most prevalent and aggressive primary brain tumor, with a median survival of only 15 months.Citation1 Standard of care treatment of GBM involves surgical resection, radiation therapy, and chemotherapy. However, there is limited long-term efficacy and inevitable tumor recurrence with this treatment approach.Citation2 Following the failure of initial treatment, second-line options for recurrent GBM are diverse, with no identified standard of care.Citation3 Immunotherapies are a promising new approach to specifically and robustly target tumor cells with reduced off-target effects, exemplified by the ability of CD19-targeting chimeric antigen receptors (CAR) T-cells to achieve durable remissions against therapy-resistant B-cell malignancies.Citation4–Citation6 Multiple clinical trials have utilized CARs to redirect T-cells against GBM-associated antigens, such as EGFRvIII and IL13 Rα.Citation7,Citation8 However, a primary challenge surrounding CAR T-cell therapy in GBM is the high variance in response rates amongst patients.Citation7 Citation7 Potential hypotheses for incomplete GBM eradication by CAR T-cells include suboptimal adoptive T-cell penetration within the tumor stroma (which can prevent the appropriate engagement and cytotoxic effects), T-cell exhaustion (which inhibits effector T-cell activity due to continuous antigen-driven stimulation), and the lack of ideal tumor-specific antigens (which can induce on-target off-site toxicities).Citation9-Citation11Currently, these limitations cannot be monitored effectively in patients until after the therapy has failed and the recurrent tumor can be analyzed from surgically resected tissue. While strategies are under investigation for systemic T-cell monitoring in patients, they are unable to image at a sufficient resolution within the tumor microenvironment to track cellular dynamics.Citation12,Citation13 Due to our inability to longitudinally monitor T-cell responses within the tumor microenvironment, there is a poor understanding of immune-based strategies to improve CAR T-cell recognition and effector function for controlling GBM progression.
Intravital fluorescence microscopy (IVM) allows for quantitative and real-time insight into T-cell therapies in preclinical mouse models.Citation14,Citation15 This in vivo imaging technique provides micron to millimeter depth of imaging in living specimens, enabling the monitoring of cell behavior within live and intact mouse brains for extended periods of time.Citation16–Citation18 A greater understanding of the spatial and temporal dynamics of the cytotoxic effects of T-cells in GBM can further elucidate the immunotherapeutic efficacy and additionally guide new areas of clinical investigation.
Using intravital microscopy, we investigated the necessity for radiation therapy (RT) in order to achieve complete tumor remission with CAR T-cells. In mouse models, complete lymphodepletion via whole-body irradiation is believed to promote CAR T-cell survival and proliferation through the upregulation and improved bioavailability of homeostatic gamma chain cytokines IL-7 and IL-15, which become available for CAR T-cell consumption without competition from endogenous lymphocytes.Citation9 While lymphodepletion appears necessary for CAR T-cell-mediated efficacy, this regimen is impractical in a clinical setting for patients with solid tumors. As such, it is important to parse out the effects of tumor-targeted RT from whole-body lymphodepletion when used synergistically with CAR T-cell therapy. Additionally, pretreating patients with RT could improve T-cell trafficking to the tumor site through enhanced immunogenicity.Citation19–Citation21
Here we report the first IVM imaging of GD2 CAR T-cells within an orthotopic GBM preclinical model. GD2 is a disialoganglioside and has been identified as a tumor antigen on neuroblastomas, sarcomas, and gliomas.Citation22–Citation24 While the GD2 CAR scFv domain has been used with human T-cells, this is the first reported use in murine T-cells in immunocompetent tumor models.Citation25 Using this approach, we longitudinally monitored the efficacy of CAR T-cells targeting the disialoganglioside GD2. We also report the first in vivo imaging of CAR T-cells inducing glioma cell death using IVM that we know of, providing insight into the mechanism of action and cell killing.
Methods
Cell lines
GL26 and SB28 murine glioma cell lines were acquired as gifts from Dr. Gerald Grant (Stanford University, Stanford, CA) and Dr. Hideho Okada (University of California San Francisco, San Francisco, CA), respectively. Culture media consisted of DMEM supplemented with 10% heat inactivated fetal bovine serum (FBS), and antibiotic-antimyocotic (ThermoFisher, Waltham, MA). Cells were maintained in a humidified, 5% CO2 incubator at 37°C. GL26-luc2/GFP cell line was generated by transfection with Lipofectamine 3000 (ThermoFisher) and three rounds of sorting for the highest 5% of GFP expressors. SB28-luc2/GFP was generated by lentiviral transduction followed by puromycin selection (125 ng/mL) and one round of sorting for the highest 5% of GFP expressors. SB28 luc2-GFP and GL26 luc2-GFP cell lines stably overexpressing GD2 were produced by with genes coding for the GD2 and GD3 synthases as described.Citation26 A stable cell line was derived through three rounds of sorting of the bulk population stained using the anti-GD2 antibody (14G2a, BioLegend, San Diego, CA) and sorted for the highest 2% of GD2 expressors. Cells were regularly tested as negative for mycoplasma by PCR and not maintained in culture for longer than 6 months.
Production of retroviral supernatant
Retroviral supernatant for the GD2 CAR was produced by transient transfection of 293GP cells with GD2.28z CAR plasmid (MSGV-14g2a-28z) and the pCL-Eco envelope plasmid. Design of the MSGV-14g2a-28z has been previously reported.Citation25 Briefly, 293GP cells were transfected via Lipofectamine 2000 (Life Technologies, Carlsbad, CA) with the plasmids encoding the CARs and the RD114 envelope protein. Supernatants were collected 48 and 72 hours after transfection.
T-cell transduction
Primary murine T-cells were isolated from spleens of healthy 6–8 week old C57Bl/6 mice (The Jackson Laboratory, Bar Harbor, ME) or transgenic mT/mG (007676, Jackson Laboratory) with cell membrane-localized tdTomato fluorescence using the EasySepTM Mouse T-cell Isolation Kit (STEMCELL Technologies, Vancouver, Canada) following manufacturer instructions. Cells were activated for 24 hours in RPMI supplemented with 10% FBS, 1% antibacterial/antimycotic solution, 50 μM 2-mercaptoethanol (ThermoFisher), 100 U/ml IL-2 (Peptrotech, Rocky Hill, NJ), 10 ng/mL IL-7 (Peprotech), and CD3/CD28 Mouse T-cell Activation DynabeadsTM (ThermoFisher) at a bead to cell ratio of 1:1. Retrovirus encoding the GD2 CAR with CD3ζ and CD28 co-stimulatory domains was centrifuged for 3 hours at 3200 RPM on non-adherent 6-well plates which had been coated overnight at 4ºC with 24 μg of RetroNectin (Takara Bio, Kusatsu, Shiga Prefecture, Japan) in 2 mL PBS per well. Viral supernatant was then removed and 1 × 106 naïve T-cells were added in 4 mL of media per well. Mock T-cells were maintained in identical activation conditions but were not transduced with the CAR vector. After 48 hours of transduction, CD3/CD28 activation beads were removed and both mock and transduced CAR T-cells were transferred to fresh medium supplemented with 100 U/ml IL-2 and 10 ng/mL IL-7. Transduction efficiencies were routinely 60-80% as measured by 1A7 staining 24 after removal of activation beads and cells were used thereafter.
T-cell functional assays
1 x 104 GL26 luc2-GFP/GD2 or SB28 luc2-GFP/GD2 tumor cells were plated per well in black 96-well plates. The following day, GD2 CAR T-cells or mock activated T-cells were added in each well at a specific effector to target ratios, ranging from 1:1 to 1:32 effector to target (312 to 1 × 104 T-cells). For cell killing assays, imaging of luciferase was performed after 24 hours of T-cell and tumor cell co-incubation on an IVIS-50 system (PerkinElmer, Waltham, MA) after addition of 56 μg D-luciferin (PerkinElmer) to each well. Images were taken with an exposure time of 30 s, f/stop of 1, and medium binning. For cytokine release assays, the supernatant was removed from individual wells and analyzed for IFN-γ, TNF-α, and IL-2 via an ELISA assay per the manufacturer instructions (Invitrogen, Carlsbad, CA).
Flow cytometry
GD2 CARs were detected with the 14g2a anti-idiotype antibody 1A7 (National Cancer Institute BRB Repository). Additional antibodies for flow cytometry include: anti-CD4 (GK1.5, BioLegend), anti-CD8a (53–6.7, BioLegend), anti-CD62 L (IM7, BioLegend), anti-CD44 (MEL-14, BioLegend), anti-GD2 (14G2a, BioLegend). Cells were washed with PBS containing 2% Bovine Serum Albumin before the addition of the antibody at a concentration of 0.2 µg per 1 × 106 cells in 100 µL volume. After 30 minutes of incubation with antibodies at 4°C in the dark, cells were washed once with PBS before being resuspended in PBS with 2% Bovine Serum Albumin (BSA). Quantitative flow cytometry was conducted using the QIFIKIT (Agilent, Santa Clara, CA). The GD2 primary antibody (14G2a, BioLegend) was added to 1 × 105 cells at a saturating dose (1 µg) and incubated at 4°C for 45 minutes. This was followed by washing and the addition of 0.5 µg of the secondary stain with APC-F(ab’)2-Goat anti-Mouse IgG (H + L) secondary antibody (Thermofisher) to samples and calibration beads. After a 30 minute incubation at 4°C, samples were washed and DAPI was added. All samples were analyzed with an LSR Fortessa or FACSAria II (BD Bioscience, San Jose, CA) and data were analyzed using FlowJo.
In vitro time-lapse imaging
Time-lapse videos were acquired using a Leica DMi8 inverted microscope with a 20x/0.40 dry objective. 1 × 105 GL26 luc2-GFP/GD2 or GL26 luc2-GFP were plated within a 6-well plate. The following day, 4 × 105 GD2 CAR T-cells were added in each well and immediately imaged at 30 second intervals. Videos were processed with ImageJ.
In vivo therapy studies
All animal experiments were performed under a protocol approved by the Stanford University Administrative Panels on Laboratory Animal Care (APLAC) and conducted in accordance with ethical guidelines prescribed therein. 6–8 week old female C57Bl/6 were implanted intracranially into the right hemisphere (0.5 mm anterior and 2 mm lateral to the lambda, 3 mm below the cranial surface) with either 1 × 104 SB28 luc2-GFP/GD2 or 1 × 105 GL26 luc2-GFP/GD2 cells in 3 µL HBSS using a Kopf stereotactic frame (David Kopf Instruments, Tujunga, CA). The cells were injected at a depth of 3 mm over a period of 7 min using an AS blunt-ended Hamilton syringe. Mice were anesthetized with 2% isofluorane over the duration of the tumor implantation and imaged with 3 mg D-luciferin 7–10 days of tumor implantation to confirm stable tumor growth. Ten days following tumor implantation, mice were randomly assigned to experimental groups based on tumor burden. Radiation treated mice were given either 5 Gy whole body irradiation (WBI) or focal RT and administered 1 × 107 CAR or mock T-cells in 100 µL of HBSS intravenously via the lateral tail vein. Tumor response was monitored by bioluminescence imaging via the IVIS 200 system (PerkinElmer). Twenty-five sequences at 30 second exposures were acquired to obtain the peak average radiance. Animals were humanely euthanized upon displaying symptoms of morbidity. Sample sizes were determined based on power calculations for one-way independent ANOVA (3 groups, power = 0.8, p < .05).
Radiation therapy
For WBI, mice were treated with 5 Gy using an x-ray cabinet (Kimtron Polaris SC-500). For focal RT, mice were treated with 5 Gy using a conformal small-animal irradiation system (X-RAD SmaRT, PXi, North Branford, CT) outfitted with a 5 mm diameter collimator. Radiation treatment planning was performed using RTImage. Cone-beam CT images were acquired prior to RT to set the radiation isocenter on the center of the tumor.
In vivo intravital imaging through a cranial window
Six to eight week old female C57Bl/6 mice were anesthetized with 2-3% isofluorine positioned on a Kopf stereotactic frame. A circular craniotomy (diameter of 3–4 mm) was performed on the right parietal bone. Tumor cells were injected as described above. Following tumor implantation, a round glass coverslip (diameter of 5 mm; Warner Instruments, Hamden, CT) was glued on the surrounding bone and further fixed to the skull by dental cement (Parkell, Edgewood, NY).
To perform intravital microscropy, mice were anesthetized with 1.5-2% isoflurane injected intravenously with 40 ug of rat anti-mouse CD31 (clone 390, BioLegend) prior to imaging. Mice were positioned on a custom-designed stereotactic frame and imaged using a Nikon confocal microscope (A1 MP+) and imaged with lasers set at 488. 560, and 633 nm. 20x water objective was selected based on the field of view and resolution considerations. Images were typically acquired over a depth of 200 µm and time-lapse videos were taken over a one-hour period.
Statistical analysis
Statistical analysis was performed using ordinary two-way ANOVA or two-tailed unpaired t tests. Statistical differences in survival curves were determined using Log-rank tests. All statistical analysis was performed in GraphPad Prism Version 8.0.2 (San Diego, CA).
Results
GD2 CAR T-cells exhibit activity against GD2 positive glioblastoma cell lines
We longitudinally monitored the efficacy of CAR T-cells targeting the disialoganglioside GD2, given it has been recently identified as a novel glioma-specific target for CAR T-cells and is under clinical investigation.Citation24 This antigen was artificially induced onto the C57BL/6 murine-derived GBM cell lines SB28 and GL26 ()). GD2 expression was quantified by flow cytometry as 1.86 ± 0.11 x 105 and 6.62 ± 0.38 x 105 molecules per cell for the SB28 GD2+ and GL26 GD2+ cell lines, respectively (Supp. Figure 1). We additionally transduced the GBM cell lines with a dual reporter gene for firefly luciferase and green fluorescent protein (GFP) for imaging with bioluminescence and fluorescence, respectively (Supp Figure 2).Citation27
Figure 1. Murine GD2 CAR T-cells are functionally active in the presence of antigen-positive tumor cells. (a) Mouse glioblastoma cell lines SB28 and GL26 were modified to express GD2 through overexpression of GD2 and GD3 synthases. SB28 GD2+ (black) and unmodified SB28 (gray) cells were stained for cell surface GD2 and analyzed by flow cytometry. (b) GD2 CAR expression was evaluated on GD2-28z (black) and untransduced mock (gray) T-cells by flow cytometry through staining with 1A7 idiotype. (c) The 14G2a single-chain variable fragment was cloned into an MSGV1 retroviral expression vector containing a CD28 transmembrane-CD28-CD3ζ signaling motif to create the MSGV.14G2a.CD28.z construct encoding the GD2 CAR. (d-k) Untransduced mock (gray) or GD2 CAR T-cells (black) were co-cultured for 24 hours with either GL26 GD2+ (d-g) or SB28 GD2+ (h-k), followed by tumor cell viability assay via bioluminescence (d,h) and ELISA of proinflammatory cytokines IFNγ, TNFα, and IL-2 (E-G, I-K). * = p < .01, ** = p < .001, *** = p < .0001 by two-way ANOVA. n = 8 per group, repeated as three independent experiments. Error bars indicate standard deviation. E:T indicates effector to target ratio.
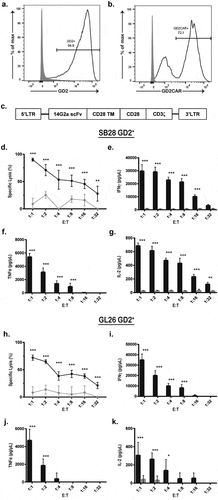
To treat the GD2-expressing gliomas, we used a GD2 CAR incorporating the scFv derived from the 14g2a antibody combined with the murine CD28 and CD3-ζ signaling domains on a MSGV1 retroviral backbone ()). Primary murine T-cells were transduced with lentivirus encoding the GD2 CAR, with subsequent flow cytometric analysis showing surface expression of GD2 at 70% (vs. <1% of mock activated T-cells) ()). Flow cytometry was used for the full characterization of GD2 CAR T-cell phenotype (Supp Figure 3). In vitro co-culture assays demonstrated substantial GD2 CAR T-cell mediated cytotoxicity against SB28 cell line overexpressing GD2 at an effector to tumor ratios of 1:1 to 1:32, with no cytotoxicity exhibited by mock activated T-cells ()). T-cell mediated cytotoxicity was further confirmed through the release of pro-inflammatory cytokines IFNγ, IL-2, and TNFα (-g)). These results were reproduced with the GL26 GD2+cell line, confirming similar cell-mediated cytotoxicity ()) and pro-inflammatory cytokine release (-k)). Lower cell killing and cytokine release were seen with the SB28 GD2+ cells compared to the GL26 GD2+ cells due to the decreased overall GD2 expression, further suggesting that CAR T-cell killing is specific to the GD2 antigen. Time-lapse microscopy visualized GD2 CAR T-cell engagement and cytotoxicity of GL26 GD2+ cells within 1 h of co-culture, confirming GD2 CAR T-cell antitumor activity (-d), Supp. Video 1).
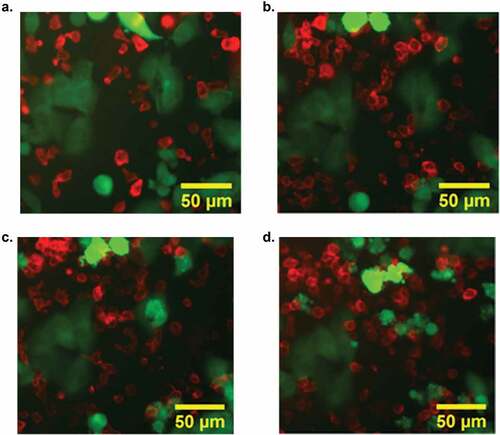
Figure 2. Confocal imaging visualized CAR T-cell (red) mediated apoptosis of tumor cells (green) within 50 minutes of cell-cell contact. GD2 CAR T-cells isolated from transgenic tdTomato mice and GL26 GFP GD2+ were plated at an effector-to-target ratio of 4:1. GD2+ tumor cells treated with GD2 CAR T-cells exhibited rapid apoptosis visualized via cell blebbing and loss of GFP signal. Image series illustrates the dynamics of CAR T-cell killing at (a) 0 minutes (b) 10 minutes (c) 15 minutes and (d) 50 minutes.
Radiation therapy with intravenous delivery of GD2 CAR T-cells induces complete GBM regression with antigen-specific T-cell memory
To evaluate in vivo efficacy of murine GD2 CAR T-cells, we treated advanced syngeneic orthotopic models of GBM with a single intravenous dose of either GD2 CAR T-cells or mock activated T-cells 1–3 hours following a sub-lethal 5 Gy whole-body irradiation (WBI) ()). In the treatment of either SB28 GD2+ or GL26 GD2+ tumors, statistically, significant improvements in antitumor response and survival were achieved with combined WBI and CAR T-cell therapy compared to CAR T-cells alone or activated nonspecific T-cell (mock) control cohorts with WBI (-d); p < .001, log rank, for both SB28 GD2+ and GL26 GD2+; n = 5–8 per group). Long-term surviving mice were re-challenged with the same GD2+ tumor in the contralateral hemisphere 10 weeks after initial treatment. Only the cohort of mice treated with both WBI and GD2 CAR T-cells suppressed tumor growth, indicating antigen-specific T-cell memory. Sustainable CAR T-cell viability was confirmed by increased circulating GD2 CAR T-cells in the periphery 3 weeks following WBI and GD2 CAR T-cell treatment compared to CAR T-cell treatment alone (), p < .01, Mann–Whitney test).
Figure 3. GD2 CAR T-cell therapy with RT effectively clears GD2+ glioblastoma in immunocompetent mouse models. (a) SB28 GD2+ or GL26 GD2+glioblastoma cells were stably transduced to express GFP and luciferase. Cells were orthotopically engrafted (1 x 105 GL26 GD2+ or SB28 GD2+) in C57BL/6 mice and were treated 10 days following tumor implantation with either 1 × 10Citation7 GD2 CAR T-cells and RT, 1 × 10Citation7 GD2 CAR T-cells alone, or 1 × 10Citation7 mock activated T-cells and RT. Both CAR T-cells and mock activated T-cells were injected intravenously. (b) Representative bioluminescent images of mice implanted with SB28 GD2+ glioblastoma cells in C57BL/6 mice with either GD2 CAR T-cells alone (left two mice) or GD2 CAR T-cells with RT (right three mice), showing tumor progression over the first 4 weeks after therapy. (c) Therapeutic monitoring of mice orthotopically implanted with SB28 GD2+ glioblastoma cells. Mice surviving 10 weeks after therapy were rechallenged with orthotopic tumors implanted on the contralateral hemisphere. n = 7 (GD2-28z + RT), 5 (GD2-28z alone), or 8 (Mock + RT). (d) Therapeutic monitoring of mice orthotopically implanted with GL26 GD2+ glioblastoma cells. Mice surviving ten weeks after therapy was rechallenged with orthotopic tumors implanted on the contralateral hemisphere. n = 11 (GD2-28z + RT), 6 (GD2-28z alone) or 6 (Mock + RT). (e) T-cell persistence was evaluated in peripheral blood lymphocytes (PBL), derived from tdTomato+ transgenic mice, administered i.v. into mice orthotopically implanted with SB28 GD2+glioblastoma cells 3 weeks following therapy. A significantly higher percentage of GD2 CAR T-cells were found in the blood of mice given both GD2 CAR T-cells and RT compared to RT alone. GD2 CAR T-cells were identified by positive staining for both 1A7 idiotype and intrinsic tdTomato signal. n = 5 (GD2-28z + RT) or 6 (GD2-28z alone). (f) Therapeutic monitoring of mice implanted orthotopically with SB28 GD2+glioblastoma cells and treated with 5 mm focal RT at 5 Gy with 1 × 10Citation7 CAR T-cells, 1 × 10Citation7 CAR T-cells alone, or 5 mm focal RT at 5 Gy with 1 × 10Citation7 mock-activated T-cells. n = 6 (GD2-28z + Focal RT), 5 (GD2-28z alone), or 7 (Focal RT alone). ** p < .001 via Log-rank test. *** p < .01 by Log-rank test. Experiments in (B), (C), and (D) were replicated twice. Error bars indicate standard error of the mean.
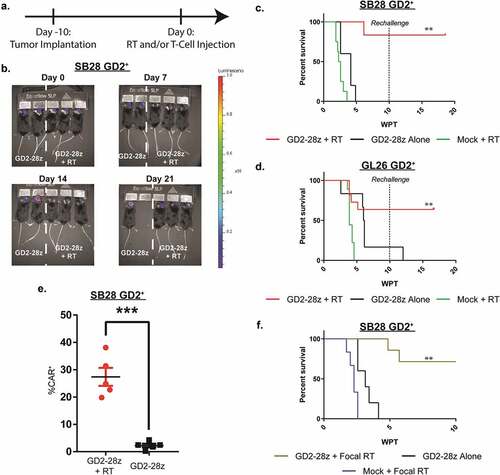
To investigate the effects of localized RT, mice were treated with 5 mm focal RT, centered at the site of tumor implantation, and GD2 CAR T-cells 10 days after implantation with SB28 GD2+ cells (Supp. Figure 4). Complete antitumor response was evident only in mice treated with the combination therapy compared to the single arm controls (); p < .001, log rank; n = 5 per group). Therapeutic response was not solely an effect of dual-arm treatment, as mice treated with GD2 CAR T-cells 4 days following WBI failed to exhibit a curative antitumor effect (Supp. Figure 5; p < .001, Log-rank).
Intravital imaging provides visualization of increased CAR T-cell extravasation and expansion within tumor microenvironment following radiation therapy
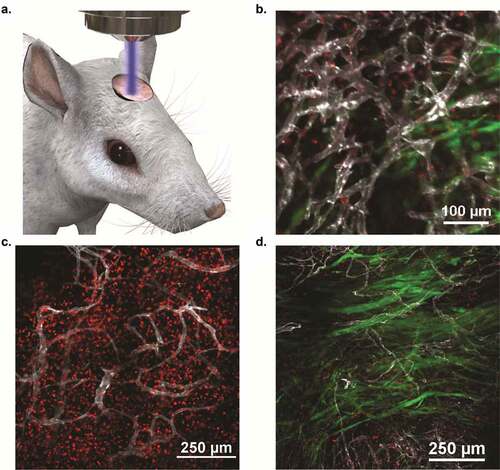
Imaging with IVM allowed for dynamic insight into the mechanisms of immune response in this preclinical mouse model ()). Failure of CAR T-cells alone was not due to loss of GD2 expression on tumor cells, as there were no significant changes in GD2 expression following RT (Supp. Figure 6A). Additionally, therapeutic failure was not due to T-cell exhaustion, as CAR T-cells isolated from a tumor site did not exhibit altered levels of exhaustion markers PD-1 and LAG-3 (Supp. Figure 6B). Therefore, longitudinal imaging within the tumor vasculature was necessary to understand the synergistic mechanisms of RT and GD2 CAR T-cells. IVM successfully visualized CAR T-cell homing and CAR T-cell-mediated apoptosis of tumor cells in real-time within the tumor vasculature in mice treated with WBI and GD2 CAR T-cells (), Supp. Video 2–3). Findings suggest that RT allows for rapid T-cell extravasation from the vasculature and local expansion within the tumor microenvironment, leading to a lasting immunologic response visualized within 5 days of treatment (), Supp. Video 4). In comparison, GD2 CAR T-cell treatment of mice without WBI resulted in insufficient extravasation within the dense tumor stroma, leading to suboptimal tumor cell access and the inability to induce complete tumor regression ()). Findings were confirmed through flow cytometry, as significantly higher levels of CAR T-cells were quantified within intracranial tumors of mice that received concurrent WBI compared to mice receiving CAR T-cells alone (Supp. Figure 7, p < .01 via Log-rank Test).
Figure 4. Intravital imaging confirmed increased CAR T-cell expansion and extravasation following RT, leading to rapid tumor clearance within 5 days. SB28 GD2+cells transfected to express GFP were orthotopically engrafted in C57BL/6 mice and were treated 10 days following tumor implantation with either 1 × 10Citation7 GD2 CAR T-cells and RT or 1 × 10Citation7 GD2 CAR T-cells alone. All adoptively transferred T-cells were derived from tdTomato mice to allow for fluorescent monitoring. (a) Visual schematic of intravital imaging shows the physical orientation of dynamic and longitudinal in vivo monitoring via a surgically implanted window chamber. (b) Intravital imaging 24 hours after treatment visualized CAR T-cell extravasation from the vasculature following whole body irradiation. (c). Intravital imaging of tumor-bearing mice 5 days following WBI and CAR T-cells revealed expansive CAR T-cell proliferation and corresponding tumor regression. (d). Imaging tumor-bearing mice 5 days following CAR T-cell treatment without WBI revealed inferior penetration within the tumor bed, leading to suboptimal therapeutic response as compared to CAR T-cells and WBI treatment. For B-D, fluorescent imaging shows SB28 GD2+ tumor cells (green), GD2 CAR T-cells (red), and tumor vasculature (white).
Discussion
Several groups have shown that CAR T-cells can induce potent antitumor responses in both immunodeficient and immunocompetent models of GBM.Citation11,Citation28-Citation30 Here we report on the necessity of RT prior to CAR T-cell therapy to induce complete tumor regression in two different immunocompetent preclinical models of GBM. In particular, we are the first to identify that the therapeutic efficacy of CAR T-cells in immunocompetent mouse models can be achieved with concurrent localized radiation. We further investigated cellular dynamics through high-resolution IVM imaging, showing RT induces CAR T-cell accumulation and extravasation throughout the tumor stroma within 24 hours of intravenous cell delivery and promotes rapid CAR T-cell proliferation within 5 days.
Many challenges exist with incorporating CAR T-cell therapy into the current treatment regimens of patients with GBM. While CAR T-cell therapies represent a promising new approach to specifically target tumor cells with minimal off-target effects, it is unclear how they will integrate with validated treatment options. With a few exceptions, most ongoing preclinical studies utilize immune-deficient mice to investigate emerging CAR T-cell options.Citation31–Citation33 While these studies can investigate the relationship between engineered human T-cells and patient-derived xenograft models, they fail to accurately represent the complexities associated with an intact immune system. They also do not to adequately assess autoimmune toxicity and the influence of host-conditioning regimens.Citation9,Citation34 Previous studies using a GD2 scFv backbone on human-derived T-cells noted CAR tonic signaling, leading to a poor overall therapeutic response.Citation25 While we did not observe tonic signaling in our model, this could be attributed to multiple tumor-associated factors, including target antigen density, growth rate, and immunosuppressive microenvironment, as well as the change from human- to murine-derived T-cells.
Studies are now beginning to utilize immunocompetent GBM models for the assessment of immunotherapies.Citation9,Citation35 These studies exclusively lymphodeplete prior to treatment, either through WBI or chemical induction, in order to maximize therapeutic efficacy, as lymphodepletion is considered a necessity for T-cell engraftment and expansion.Citation36 However, the necessity of lymphodepletion in preclinical immunocompetent GBM studies has not been fully understood prior to the current study. Our study suggests that full lymphodepletion is not required, as focal RT at the tumor site along with CAR T-cells is sufficient to induce complete tumor regression. Previous studies indicate local fractionated radiotherapy can increase T-cell infiltration, with the effects failing to extend beyond the treatment site.Citation20 Additionally, radiation-induced degradation of the extracellular matrix can enhance T-cell migration and engagement within the tumor stroma.Citation37–Citation39 Introducing an unnatural antigen to the GBM cells had the potential to induce an innate immune response, which would be enhanced upon radiation therapy through increased antigen bioavailability upon tumor cell death. However, radiation therapy alone was unable to induce a complete tumor remission in these models, indicating the introduction of a foreign antigen did not induce a significant immune response.
This approach has high translational potential by incorporating stereotactic radiosurgery into clinical trials evaluating CAR-T-cell therapy in GBM patients. While challenges exist in the maximal tolerated radiation dosage within the brain, retreatment with RT is feasible in certain patients and can lead to improved overall survival.Citation40–Citation42 Additionally, innovations in PET-guided RT can utilize metabolic imaging to strategically deliver higher radiation through dose painting, minimizing off-site toxicities.Citation43,Citation44 Pretreatment of patients with targeted RT before CAR T-cells infusion should be investigated in clinical trials.
We have established an imaging methodology to study the progression of orthotopically implanted GBM cells in response to treatment, in this case, CAR T-cell therapy and RT in an immunocompetent preclinical GBM model. This approach can be extended to other GBM cell lines or adoptive T-cell models, provided both cell populations stably express fluorescent reporters. While we are limited to imaging only the superficial layers of the cortex (upwards of 250 um), this imaging technique has the advantage of enabling longitudinal imaging of cellular response and vascular remodeling with high resolution. While the slight drift of the mouse during the imaging timeframe limited our ability to generate quantitative metrics on acquired images, a more robust and stabilizing imaging rig would allow for more accurate measurements of CAR T-cell kinetics within the tumor microenvironment. Imaging can differentiate serial CAR T-cell killing, where a single CAR T-cell induces apoptosis of multiple tumor cells, versus rapid CAR T-cell proliferation to induce tumor cell death. Future studies can utilize this imaging technique to investigate antigen escape through implantation of both GD2+ and GD2− tumor cells, providing further insight into the patient treatment of GBM with CAR T-cells. Additionally, investigations into engineering CAR T-cells for enhanced extravasation from the neovasculature into the tumor stroma will lead to improved therapeutic outcomes.
Authors contributions
S.M., S.T.H., C.L.M, and S.S.G conceived of the project and designed all experiments. S.M., S.T.H., C.B., A.A., I.S.A, T.M.S., C.B.P. and E.E.G performed experiments. S.M., S.T.H., and T.M. contributed to data analysis. S.M. and S.S.G. wrote the manuscript with contributions from all authors.
Supplemental Material
Download ()Acknowledgments
The authors would like to acknowledge the Stanford Center for Innovation in In-Vivo Imaging (SCI3) and, in particular, Timothy Doyle for their maintenance of the preclinical animal imaging facility. We also thank members of the Stanford FACS facility for their expertise. We thank Dr. Seung-Min Park for his assistance in the illustrations. Funding for this work was provided by the Canary Foundation (S.S.G.) and the Ben and Catherine Ivy Foundation (S.S.G.). Data were collected on an instrument in the Shared FACS Facility obtained using NIH S10 Shared Instrument Grant S10RR027431-01. S.M. has received support from the National Science Foundation Graduate Student Fellowship.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Johnson DR, O’Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol. 2012;107(2):359–10. doi:10.1007/s11060-011-0749-4.
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med [Internet]. 2005;352(10):987–996. http://login.proxy.library.vanderbilt.edu/login?url=http://search.proquest.com/docview/223934687?accountid=14816%5Cnhttp://sfx.library.vanderbilt.edu/vu?url_ver=Z39.88-2004&rft_val_fmt=info:ofi/fmt:kev:mtx:journal&genre=article&sid=ProQ:ProQ:healthcomplet.
- Gallego O. Nonsurgical treatment of recurrent glioblastoma. Curr Oncol [Internet]. 2015;22(4):e273–81. doi:10.3747/co.22.2436.
- Lin Y, Okada H. Cellular immunotherapy for malignant gliomas. Expert Opin Biol Ther [Internet]. 2016;2598(August):1–11. doi:10.1080/14712598.2016.1214266.
- Galluzzi L, Vacchelli E, Bravo-San Pedro J-M, Buqué A, Senovilla L, Baracco EE, Bloy N, Castoldi F, Abastado J-P, Agostinis P, et al. Classification of current anticancer immunotherapies. Oncotarget. 2014;5(24). doi:10.18632/oncotarget.2998.
- Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, Guo X, Liu H, Ding N, Zhang T, et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med [Internet]. 2019;25(86):947–953. http://www.nature.com/articles/s41591-019-0421-7.
- O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(July):eaaa0984. doi:10.1126/scitranslmed.aaa0984.
- Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med [Internet]. 2016;375(26):2561–2569. http://www.ncbi.nlm.nih.gov/pubmed/28029927%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC5390684.
- Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, Schmittling RJ, Nair SK, Reap EA, Norberg PK, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. 2014;20(4):972–984. doi:10.1158/1078-0432.CCR-13-0709.
- Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, Nace AK, Dentchev T, Thekkat P, Loew A, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med [Internet]. 2015;7(275):275ra22–275ra22. doi:10.1126/scitranslmed.aaa4963.
- Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, Wakefield A, Fousek K, Bielamowicz K, Chow KKH, et al. Tandem CAR T cells targeting HER2 and IL13Ra2 mitigate tumor antigen escape. J Clin Invest. 2016;126(8):3036–3052. doi:10.1172/JCI83416.
- Keu KV, Witney TH, Yaghoubi S, Rosenberg J, Kurien A, Magnusson R, Williams J, Habte F, Wagner JR, Forman S, et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci Transl Med [Internet]. 2017;9(373). http://stm.sciencemag.org/lookup/doi/10.1126/scitranslmed.aag2196%5Cnpapers3://publication/doi/10.1126/scitranslmed.aag2196.
- Tavaré R, McCracken MN, Zettlitz KA, Knowles SM, Salazar FB, Olafsen T, Witte ON, Wu AM. Engineered antibody fragments for immuno-PET imaging of endogenous CD8+ T cells in vivo. Proc Natl Acad Sci U S A [Internet]. 2014;111(3):1108–1113. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3903195&tool=pmcentrez&rendertype=abstract.
- Boissonnas A, Fetler L, Zeelenberg IS, Hugues S, Amigorena S. In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J Exp Med [Internet]. 2007;204(2):345–356. http://www.ncbi.nlm.nih.gov/pubmed/17261634%5Cnhttp://jem.rupress.org/content/204/2/345.full.pdf%5Cn%3CGotoISI%3E://000244504100014%5Cnhttp://www.ncbi.nlm.nih.gov/pmc/articles/PMC2118741/pdf/jem2040345.pdf.
- Lodygin D, Odoardi F, Schläger C, Körner H, Kitz A, Nosov M, van den Brandt J, Reichardt HM, Haberl M, Flügel A, et al. A combination of fluorescent NFAT and H2B sensors uncovers dynamics of T cell activation in real time during CNS autoimmunity. Nat Med [Internet]. 2013;19(6):784–790. http://www.ncbi.nlm.nih.gov/pubmed/23624600.
- Ricard C, Debarbieux FC. Six-color intravital two-photon imaging of brain tumors and their dynamic microenvironment. Front Cell Neurosci [Internet]. 2014;8(February):57. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3932518&tool=pmcentrez&rendertype=abstract.
- Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WEF, Goldbrunner R, Herms J, Winkler F. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med [Internet]. 2010;16(1):116–122. http://www.ncbi.nlm.nih.gov/pubmed/20023634.
- Bayerl SH, Niesner R, Cseresnyes Z, Radbruch H, Pohlan J, Brandenburg S, Czabanka MA, Vajkoczy P. Time lapse In Vivo microscopy reveals distinct dynamics of microglia-tumor environment interactions — a new role for the tumor perivascular space as highway for trafficking microglia. Glia. 2016.
- Spary LK, Al-Taei S, Salimu J, Cook AD, Ager A, Watson HA, Clayton A, Staffurth J, Mason MD, Tabi Z. Enhancement of T cell responses as a result of synergy between lower doses of radiation and T cell stimulation; 2019.
- Dovedi SJ, Cheadle EJ, Popple AL, Poon E, Morrow M, Stewart R, Yusko EC, Sanders CM, Vignali M, Emerson RO, et al. Fractionated radiation therapy stimulates antitumor immunity mediated by both resident and infiltrating polyclonal T-cell populations when combined with PD-1 blockade. J Immunol. 2017;23(18):5514–5527.
- Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, Camphausen K, Luiten RM, de Ru AH, Neijssen J, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203(5):1259–1271. doi:10.1084/jem.20052494.
- Schulz G, Cheresh DA, Varki NM, Yu A, Staffileno LK, Reisfeld RA. Detection of ganglioside GD2 in tumor tissues and sera of neuroblastoma. Cancer Res. 1984;44:5914–5920.
- Dobrenkov K, Cheung NV, Ostrovnaya I, Gu J, Cheung IY. Oncotargets GD2 and GD3 are highly expressed in sarcomas of children, adolescents, and young adults. Pediatr Blood Cancer. 2016;63(10):1780–1785. doi:10.1002/pbc.26097.
- Mount CW, Majzner RG, Sundaresh S, Arnold EP, Kadapakkam M, Haile S, Labanieh L, Hulleman E, Woo PJ, Rietberg SP, et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat Med [Internet]. 2018;24(May). http://www.nature.com/articles/s41591-018-0006-x.
- Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med [Internet]. 2015;21(6):581–590. http://www.ncbi.nlm.nih.gov/pubmed/25939063.
- Haraguchi M, Yamashiro S, Furukawa K, Furukawa K, Takamiya K, Lloyd KO, Shiku H, Furukawa K. Isolation of GD3 synthase gene by expression cloning of GM3 a-2,8-sialyltransferase cDNA using anti-GD2 monoclonal antibody. Proc Natl Acad Sci U S A. 1994;91(October):10455–10459. doi:10.1073/pnas.91.22.10455.
- Ray P, Gambhir SS. Noninvasive imaging of molecular events with bioluminescent reporter genes in living subjects. Methods Mol Bio. 2007.
- Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, Rainusso N, Wu M-F, Liu H, Kew Y, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther [Internet]. 2013;21(3):629–637. http://linkinghub.elsevier.com/retrieve/pii/S1525001616306323.
- Kong S, Sengupta S, Tyler B, Bais AJ, Ma Q, Doucette S, Zhou J, Sahin A, Carter BS, Brem H, et al. Suppression of human glioma xenografts with second-generation IL13R-specific chimeric antigen receptor-modified T cells. Clin Cancer Res. 2012;18(21):5949–5960. doi:10.1158/1078-0432.CCR-12-0319.
- Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-based CAR T cells and radiotherapy exert synergistic efficacy in glioblastoma. Cancer Res. 2018;78(4):1031–1043. doi:10.1158/0008-5472.CAN-17-1788.
- Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, Grossman RG, Heslop HE, Gottschalk S. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16(2):474–485. doi:10.1158/1078-0432.CCR-09-1322.
- Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64(24):9160–9166. doi:10.1158/0008-5472.CAN-04-0454.
- Liu K, Liu X, Peng Z, Sun H, Zhang M, Zhang J, Liu S, Hao L, Lu G, Zheng K, et al. Retargeted human avidin-CAR T cells for adoptive immunotherapy of EGFRvIII expressing gliomas and their evaluation via optical imaging. Oncotarget [Internet]. 2015;6(27):23735–23747. http://www.ncbi.nlm.nih.gov/pubmed/26124178.
- Richman SA, Nunez-Cruz S, Moghimi B, Li LZ, Gershenson ZT, Mourelatos Z, Barrett DM, Grupp SA, Milone MC. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol Res [Internet]. 2017;(Mcm):canimm.0211.2017. doi:10.1158/2326-6066.CIR-17-0211.
- Flores C, Pham C, Snyder D, Yang S, Sanchez-Perez L, Sayour E, Cui X, Kemeny H, Friedman H, Bigner DD, et al. Novel role of hematopoietic stem cells in immunologic rejection of malignant gliomas. Oncoimmunology [Internet]. 2015;4(3):e994374. doi:10.4161/2162402X.2014.994374.
- Wang LX, Shu S, Plautz GE. Host lymphodepletion augments T cell adoptive immunotherapy through enhanced intratumoral proliferation of effector cells. Cancer Res. 2005;65(20):9547–9554. doi:10.1158/0008-5472.CAN-05-1175.
- Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science (80-). 2015;348(6230):6230. doi:10.1126/science.aaa6204.
- Salmon H, Franciszkiewicz K, Damotte D, Validire P, Trautmann A, Mami-chouaib F, Mami-Chouaib F, Donnadieu E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. 2012;122(3):899–910. doi:10.1172/JCI45817.
- Murty S, Gilliland T, Qiao P, Tabtieng T, Higbee E, Al Zaki A, Puré E, Tsourkas A. Nanoparticles functionalized with collagenase exhibit improved tumor accumulation in a murine xenograft model. Part Part Syst Charact [Internet]. 2014 Nov 10 [cited 2014 Nov 20];n/a-n/a. doi:10.1002/ppsc.201400169.
- Krauze A, Attia A, Braunstein S, Chan M, Combs S, Fietkau R. Expert consensus on re-irradiation for recurrent glioma. 2017;1–10.
- Amelio D, Amichetti MA. Radiation therapy for the treatment of recurrent glioblastoma: an overview. Cancers (Basel). 2012;4(1):257–280. doi:10.3390/cancers4010257.
- Kazmi F, Yang Y, Yiat S, Leong H, Yao W, Balamurugan K. Re-irradiation for recurrent glioblastoma (GBM): a systematic review and meta-analysis. J Neurooncol [Internet]. 2019;142(1):79–90. doi:10.1007/s11060-018-03064-0.
- Shah JL, Li G, Shaffer JL, Azoulay MI, Gibbs IC, Nagpal S, Soltys SG. Stereotactic radiosurgery and hypofractionated. Neurosurgery. 2018;82(1):24–34. doi:10.1093/neuros/nyx115.
- Menichetti L, Petroni D, Panetta D, Burchielli S, Bortolussi S, Matteucci M, Pascali G, Del Turco S, Del Guerra A, Altieri S, et al. A micro-PET/CT approach using O-(2-[18F]fluoroethyl)-l-tyrosine in an experimental animal model of F98 glioma for BNCT. Appl Radiat Isot [Internet]. 2011;69(12):1717–1720. doi:10.1016/j.apradiso.2011.02.037.