
ABSTRACT
Clinical observations suggest that responses to cancer immunotherapy are correlated with intra-tumoral T cell receptor (TCR) clonality, tumor mutation burden (TMB) and host HLA genotype, highlighting the importance of host T cell recognition of tumor antigens. However, the dynamic interplay between T cell activation state and changes in TCR repertoire in driving the identification of potential immunodominant antigen(s) remains largely unexplored. Here, we performed single-cell RNA-sequencing on CD8+ tumor-infiltrating T cells (TILs) using the murine colorectal tumor model MC38 to identify unique TCR sequences and validate their tumor reactivity. We found that the majority of clonally expanded TILs are tumor-reactive and their TCR repertoire is unique amongst individual MC38 tumor-bearing mice. Our query identified that multiple expanded TCR clones recognized the retroviral epitope p15E as an immunodominant antigen. In addition, we found that the endogenous retroviral genome encoding for p15E is highly expressed in MC38 tumors, but not in normal tissues, due to epigenetic derepression. Further, we demonstrated that the p15E-specific TILs exhibit an activated phenotype and an increase in frequency upon treatment with anti-41BB and anti-PD-1 combination immunotherapy. Importantly, we showed that although p15E-specific TILs are not required to mount a primary anti-tumor response, they contributed to the development of strong immune memory. Overall our results revealed that endogenous retroviral antigens expressed by tumor cells may represent an important and underappreciated category of tumor antigens that could be readily targeted in the clinic.
Introduction
Immunotherapy has revolutionized cancer treatment. However, the response rate to existing immune-modulatory agents across multiple tumor types remains low. Considerable efforts are ongoing to identify patients who are more likely to respond to checkpoint inhibition through the identification of predictive biomarkers.Citation1 Intra-tumoral T-cell receptor (TCR) clonality, tumor microsatellite burden (TMB), and host HLA genotype are associated with responses to immunotherapy, which highlights the importance of host T cell recognition of tumor antigens.Citation2–Citation4 Indeed, the analysis of the TILs phenotype and TCR clonality, together with the identification of the corresponding tumor antigens may help elucidate the mechanism of the response to current immunotherapies and facilitate the development of novel therapeutics.
Specific antigens are recognized by unique T cell receptors and thus monitoring the TCR repertoire changes in periphery and tumors during immunotherapies helps directly evaluate T cell response.Citation1 TCRs are generated through V(D)J recombination during thymic development to ensure sufficient diversity for the recognition of an infinite number of pathogens.Citation5 However, the enormous size and diversity of the TCR repertoire make it challenging to study. Historically, various methods have been used to estimate the TCR repertoire, including TCR-Vβ usage, immunoscope (TCR-Vβ in combination with CDR3 length), bulk RNA-seq, or DNA-seq.Citation6 Recently, the development of single-cell sequencing enables us to obtain both the TCR alpha and beta sequences together with transcriptome information at the single-cell level.Citation7 Although some investigational clinical studies using single-cell sequencing have revealed many important aspects of native TCR repertoire, Citation7,Citation8 many studies still use bulk DNA sequencing for TCR repertoire assessments since single-cell sequencing demands timely preparation of fresh tumor samples.Citation3,Citation9
Further, studies linking TCR clonality and tumor antigen reactivity are limited. Previous tumor reactivity studies were based on a co-culture of TILs and tumor cells to generate oligo-clonal tumor-reactive T cells for autologous transfer in cancer patients and identification of their corresponding tumor antigens.Citation10,Citation11 However, the long-term co-culture process alters the original phenotype and TCR distribution of these T cells. With the advance of single-cell sequencing as well as pairing algorithms, paired TCR sequences can be obtained, cloned, and tested for reactivity. It has been shown that CD8+ intratumoral TCR repertoire has low and variable tumor reactivity in human tumor types, such as high-grade serous ovarian cancer and microsatellite stable colorectal cancer carcinoma, with low response rate to PD-1 blockade.Citation12 In addition, TCRs from intratumoral CD4+ regulatory T cells were shown to recognize neoantigens and are indeed tumor reactive.Citation13 TCRs reactive to murine melanoma B16 were also identified through single-cell TCR sequencing of 4-1BB+CD8TILs.Citation14
Due to the enormous diversity of both TCRs and antigens, identification of the corresponding antigens for tumor-specific TCRs poises a technical challenge. Previous efforts to identify tumor antigens relied heavily on existing host immunity, which starts from obtaining patient tumor-specific cytotoxic T lymphocytes (CTLs) and sera.Citation10 By screening cDNA libraries, Citation15,Citation16 eluted peptide libraries,Citation17 and most recently synthetic peptide-HLA libraries, Citation18 many well-known tumor antigens were identified, such as MAGE family, Tyrosinase, NY-ESO1, and P-MEL. Current in-silicon prediction algorithms with tumor mass spectrometry and exome sequencing data are also able to efficiently predict neo-epitopes in murine tumors.Citation19,Citation20 The prediction algorithms have assisted personalized tumor vaccines which yielded robust anti-tumor efficacy in clinical settings.Citation21 On the other hand, in-silicon antigen prediction algorithms based on TCR sequences are still limited to particular antigen categories and their accuracy remains to be proven.Citation22 Overall, better understanding of tumor antigens, including their quantity, quality, and origin, will allow a more accurate estimation of tumor immunogenicity.
In this study, we sought to investigate the tumor reactivity and clonality of the intratumoral TCR repertoire and identify its corresponding antigens, as well as to further link to in situ T cell phenotype. To that end, we performed for the first time a comprehensive analysis combining single-cell RNA sequencing, in-silicon antigen prediction, TCR cloning, and in vitro functional testing. We applied this analysis on CD8+ TILs from murine MC38 tumor model, due to their central role in the development of anti-tumor immunity.Citation23–Citation25 We found that the majority of clonally expanded TILs were tumor-reactive and the TCR repertoire is unique amongst individual MC38 tumor-bearing mice. We identified the endogenous retroviral epitope p15E as an immunodominant antigen and revealed that the ectopic expression of the endogenous viral genome in the tumor is due to epigenetic de-repression. These findings were validated in vivo by demonstrating that p15E-specific TILs exhibit an activated phenotype and expand upon anti-PD-1 and anti-41BB combination immunotherapy. Genetic inactivation of p15E from MC38 did not affect the response to combination immunotherapy, but it did impair the induction of immune memory. We conclude that endogenous retroviral antigens represent an important category of tumor antigens in murine tumors, which may also be relevant in clinical settings.
Materials and methods
Animal studies
All procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals of the NIH. The protocols were approved by the Regeneron Pharmaceuticals Institutional Animal Care and Use Committee (IACUC).
Cell lines
MC38 (NIH), B16F1 (ATCC-CRL6323), B16F10 (ATCC-CRL6475), B16F10.9 (a subline from B16F10), EL4 (ATCC-TIB-39), J.RT3-T3.5 (abbreviated as JRT) (ATCC-TIB-153) and TrampC2 (ATCC-CRL2731) were cultured according to ATCC recommended protocols. All cells were cultured at 37°C 5% CO2 unless indicated otherwise. Briefly, MC38 was cultured in DMEM + 10% FBS + Pen/Strep/Glutamine (abbreviated as P/S/G hereafter) + NaPyr + NEAA. B16F1, B16F10, B16F10.9 were cultured in DMEM + 10% FBS + P/S/G. TrampC2 was cultured with DMEM + 5% FBS + 5% NuSerum IV + P/S/G + 0.005 mg/ml insulin + 10 nM Dihydrotestosterone (DHT). EL4 was cultured with RPMI1640 + 10% FBS + P/S/G. JRT reporter cell lines were generated from JRT cell line by lentiviral transduction of AP1-Luc, mCD8α/β and mCD28. JRT reporter cells were cultured with RPMI1640 + 10% FBS + P/S/G + 1μg/mL Puro + 500μg/mL Neo. JRT reporter cells were later transduced with different TCRs and were cultured with RPMI1640 + 10% FBS + P/S/G + 1μg/mL Puro + 500μg/mL Neo + 200μg/mL Zeocin.
JRT-TCR cell line generation
From the single-cell RNA sequencing results, mouse TCR alpha and beta sequences with codon-optimized variable regions were assembled and cloned into lentiviral vector pLVX-EF1a-Zeocin (Clontech) downstream of EF1a promoter, followed by human TCR alpha and beta constant sequences, respectively. Chimeric TCR alpha and beta chains were linked by Furin-2A element to ensure equimolar ratio between alpha and beta chains. HEK293 cells were transiently transfected with pLVX plasmid containing TCR sequences, psPAX2 and pMD2 G by Lipofectamine 3000 (Thermo Fisher). The produced lentivirus was used to generating JRT reporter cell line with stable surface expression of chimeric TCR receptors. TCR-transduced JRT cells underwent Zeocin selection and fluorescent-activated cell sorting, yielding >95% purity.
JRT-TCR reporter assay
To test the TCR reactivity, 0.5 × 106 TCR-transduced JRT cells were incubated with desired number of target cells at 37°C 5% CO2 for 5 hours. In some experiments, target cells were treated with 100 ng/ml recombinant mIFNγ (R&D system 485-MI) overnight before co-culturing with JRT cells. Cells were lysed and luciferase activity was stimulated using ONE-Glo Luciferase assay system (Promega). Briefly, 100 μl ONE-Glo reagent was added to each well and mixed with cells. After 5 minutes of incubation at room temperature, luciferase activity was assessed by SpectraMax M5 plate reader (Molecular Probes). To eliminate the differences of TCR expression levels among different JRT cell lines, relative luminescence unit (RLU) was normalized to the condition with Human T-Activator DynaBeads at 2:1 bead-to-cell ratio (Thermo Fisher).
Antibodies and reagents
Antibodies for flow cytometry and FACS: H2Kb PECy7 (eBioscience 25-5958-82), H2Db PE (BioLegend 111508), I-A/I-E (MHCII) BV711 (BD Biosciences 563414), PDL1 BV41 (BD Biosciences 564716), CD8 APC-eFluor 780 (eBioscience 47-0081-82), DAPI (Sigma, D9564-10 MG), PD1 PerCP-eFluor710 (Invitrogen 46-9985-82), CD44 BUV737 (BD Biosciences 564392), CD62 L BV786 (BD Biosciences 564109), hTCRab PerCP-Cy5.5 (Biolegend 306724), hCD3 PECy7 (BD Biosciences 563423). p15E (KSPWFTTL), OVA (SIINFEKL) peptides and corresponding pentamers were from ProImmune.
C1 fluidigm single-cell sequencing
Single-cell sequencing method was described previously.Citation26 Briefly, 1 × 106 MC38 cells were implanted on the flank of C57BL/6 mice. Tumor sizes were measured twice per week using calipers (Roboz RS-6466). Tumor volume was calculated using the formula W^2 × L × 0.5, where L is the longest dimension and W is the perpendicular dimension. Mice with tumors larger than 2000 mm3 or with ulceration were euthanized by CO2. The mice were euthanized by CO2 14 days post-implantation before tumors and spleen were harvested. Tumors and spleen were processed using Miltenyi gentleMACS® Octo dissociator and mouse tumor dissociation kit (130-096-730) according to manufacturer’s instructions. Cells were stained with anti-CD8 antibody APC-eFluor 780 (eBioscience 47-0081-82) and DAPI (Sigma, D9564-10 MG). CD8 positive live single cells were sorted by using Beckman Coulter MoFlo® Astrios.
Single-cell sequencing was performed on C1 fluidigm platform according to the manufacturer’s instruction. Cells are prepared at a concentration of 200–500 cells/μl in the favorable medium, and then mixed with C1 Cell Suspension Reagent (Fluidigm) at the ratio of 3:2 and loaded onto C1 IFC (10–17 um). The LIVE/DEAD staining solution (2.5 μl ethidium homodimer-1 and 0.625 μl calcein AM (Life Tech mp03224) added to 1.25 ml C1 Cell Wash Buffer) is loaded onto C1 IFC for determining the live/dead status of cells. Each capture site is examined by Nikon microscope/Leica DMI8 microscope in the brightfield, GFP, and TexasRed channels for the number of cells and live/dead status of cells. Cell lysing, reverse transcription, and mRNA amplification were performed on the C1 Single-Cell Auto Prep Integrated Fluidic Circuit (IFC) following the methods described in the protocol (100–7168 E1). The SMARTer Ultra Low RNA Kit (Clontech 634833, 1 kit for 10 C1 IFCs) is used for cDNA synthesis from the single cells. On the second day, remove 3 μl of cDNA from C1 IFC and add into 10 μl DNA Dilution Buffer (Fluidigm). cDNA is quantitated by Qubit High Sensitivity dsDNA assay (Life Tech, Q32854) on the Tecan plate reader (Tecan). Illumina NGS library construction uses Nextera XT Sample Prep kit (Illumina) by following the instruction in the protocol (100–7168 E1). Sequencing was performed on Illumina HiSeq®2500 (Illumina) rapid mode by multiplexed single-read run with 50 cycles. Decoding was run by Casava (illumina). Mapping was done using CLC bio Genomics Workbench Version 7.0 (CLC Bio).
p15E knockout MC38 cell line generation and validation
MC38WT single-cloned cell line was first generated from parental MC38 cell line by single-cell FACS sorting. sgRNA targeting p15E (KSPWFTTL) coding sequence was designed using sgRNA designer from BROAD institute (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). A total of 1μg of NLS-Cas9-EGFP nuclease (Genscript) and 240 ng of sgRNA (5ʹ-TATCAGGGTGGTGAACCAAG-3ʹ from Synthego) were mixed in 5 μl resuspension buffer R at room temperature for 10 minutes to form Cas9-sgRNA complex. A total of 0.5 × 106 MC38WT cells in 5 μl resuspension buffer R were mixed with 5 μl Cas9-sgRNA complex and were electroporated with Neon transfection system at 1550 V 10 ms for 3 pulses (ThermoFisher). MC38 knockout single-cloned cell lines were generated by single-cell FACS sorting and later subjected to JRT luciferase assay screening. Single-cell clones tested negative for JRT luciferase assay were subjected to Amplicon sequencing (Amplicon-EZ service from Genwiz). Single-cell clones were digested by protein K (Qiagen) and lysates were used as template to amplify p15E-coding region with forward primer (5ʹ-TTTGACCTCCTTGTCCGAAG-3ʹ) and reverse primer (5ʹ-CTCA ATCGCCTGGTCCAGTT-3ʹ) from IDT. Unique DNA and amino acid sequences were aligned to wildtype genomic sequences (Supplementary Figure 3). The clone with complete disruptions of original p15E DNA and amino acid sequences was deemed as MC38KO cell line.
Mouse primary T cell in vitro culture and IFNγ ELISPOT
MC38 tumors and spleens were harvested and processed 37 days post implantation. Single-cell suspensions from six tumors were made by Miltenyi Mouse tumor dissociation kit (130-096-730). Single-cell suspensions were later pooled and cultured in vitro briefly. The cells were further purified by Percoll (40/80) and CD8TIL purification kit (Miltenyi 130-116-478) before FACS sorting. Primary T cells were later expanded in vitro with CD3/CD28 DynaBeads in T cell culture media (RPMI1640 + 10% FBS + P/S/G + NaPyr + NEAA + 2-Mercaptoethanol) with 100 U/mL IL2 and 2 ng/mL IL7 for 12 days. IFNγ ELISPOT was performed with mouse IFNγ ELISPOT set (BD Biosciences 551083). A total of 5 × 103 T cells and 1 × 105 corresponding tumor cells were mixed and incubated overnight. Plates were processed according to manufacturer's instruction and TMB substrate (MABTECH) was used to develop the spots. Plates were scanned on AID iSPOT reader and enzyme activity was calculated by Elispot7.0 iSpot software. Specific cytokine release activity was calculated by subtracting background cytokine release activity of TrampC2 group.
DNA methylation sequencing
Genomic DNA was extracted from MC38 cells, TrampC2 cells, and C57BL/6 spleen tissue using blood & cell culture DNA mini kit (Qiagen). A total of 500 ng of genomic DNA was used as input for sodium bisulfite conversion with EZ DNA Methylation-Lightning kit (Zymo Research). Modified DNA was then amplified by forward primer REGNMP379 (5ʹ-CTACACGACGC TCTTCCGATCTAGTTGGGTAGTTAATTATTTTGAGGAG-3ʹ) and reverse primer REGNMP380 (5ʹ-CAGACGTGTGCTCTTCCGATCTACTATCTCAAAATTCCCCAAATAA C-3ʹ), then 1st round PCR product was purified via AmpPure XP Beads (Beckman Coulter). The purified 1st round PCR product was amplified by primers OEM14 (5ʹ- CAGACGTGTGCTC TTCCGATCTACTATCTCAAAATTCCCCAAATAAC-3ʹ) and REGN barcoded primer (5ʹ- CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3ʹ, XXXXXXXX is an 8-bp barcode for NGS libraries), then purified again with AmPure XP Beads (Beckman Coulter). Sequencing of the PCR products was performed on Illumina MiSeq (Illumina) by multiplexed paired-read run with 150 cycles (pair ends 150 bp).
Exome-sequencing and neoantigen alignment
Genomics DNA (gDNA) was purified from about one million frozen cells using Qiagen Tissue and Blood DNA Extraction Kit (Qiagen). The concentration of gDNA was determined using Qubit dsDNA Broad Range assay by Qubit 2.0 Fluorometer (Life Technologies). DNA-seq libraries were prepared from 1 µg gDNA using KAPA HyperPrep Kit (KAPA Biosystems). Exome capture was performed following the procedure of Agilent SureSelect platform (SureSelectXT2 Mouse All Exon). Sequencing was performed on Illumina HiSeq®2500 (Illumina) by multiplexed paired-read run with 2 × 100 cycles. Reads were mapped to reference genome and regions containing neoantigens were identified according to the publications.Citation20 Translated amino acid sequences were compared to reported neoantigens and their presence in our in-house cell line was determined.
RNA-sequencing, p15E coding sequence alignment, and proteasome gene expression comparison
Strand-specific RNA-seq libraries were prepared from 1 µg RNA using KAPA stranded mRNA-Seq Kit (KAPA Biosystems). Twelve-cycle PCR was performed to amplify libraries. The amplified libraries were size-selected at 400–600 bp using PippinHT (Sage Science). Sequencing was performed on Illumina HiSeq®2500 (Illumina) by multiplexed paired-read run with 2 × 100 cycles. Reads were mapped to reference genome. p15E coding region was aligned across different cell lines. Gene expression of proteasome genes was extracted by using OmniSoft® Array Studio software. Hierarchical clustering among proteasome genes was performed across different cell lines.
Results
Expanded TCR clones were detected in mouse MC38 tumors using single-cell RNA sequencing
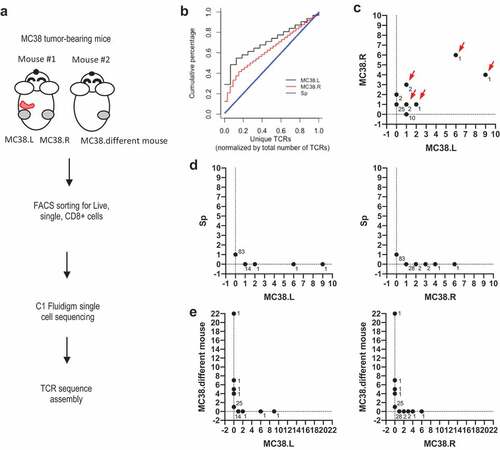
It has been previously shown that CD8+ TILs are essential in anti-tumor immunity as CD8+ depletion greatly affected the anti-tumor effects of different cancer immunotherapy treatments in mice.Citation23–Citation25,Citation27 To determine the diversity and TCR clonality of antigen-specific intratumoral CD8+ T cells, we selected a widely used syngeneic murine colon carcinoma tumor model MC38 derived in C57BL/6 background. We examined if TCR repertoires were similar for the same tumor cell line implanted at different anatomical locations from the same mouse (left vs right flank). In addition, we studied TCR repertoires from the same tumor cell line implanted in different mice to assess how naïve TCR repertoires influence tumor TCR repertoires and to examine the existence of “public” TCRs specific for MC38. We implanted MC38 cells subcutaneously on the left and right flanks of C57/BL6 mice (schematic of experimental design shown in ). Fourteen days post implantation, tumor tissues and spleen were harvested and processed into single-cell suspension. CD8+ TILs were purified by fluorescence-activated cell sorting (FACS) before transcriptome and TCR profiling. TCR sequences were assembled from short reads as previously described and unique TCRs were defined as unique TCRβ CDR3 amino acid sequences.Citation27 To quantify and compare the relative frequency of expanded TCR clones found in the tumors and spleens, we generated cumulative curves of normalized unique TCRs. We found that the TCRs from the spleen were unique and lack of clonal expansion (linear curve) (). In contrast, tumor TCRs showed increased clonal expansion relative to the spleen (). Further, several high-frequency TCR clones were shared amongst TIL cells isolated from MC38 tumors implanted at different sites on an individual mouse (highlighted by red arrows in ). However, there were no shared TCRs in MC38 tumors and spleens from the same mouse, suggesting that the TILs specifically expanded and/or accumulated at the tumor sites (). Surprisingly, MC38 tumors from different mice did not share any TCR clones (). This data suggests that expanded TCRs are likely tumor-specific but are unique to individual mice. This difference amongst individual mice is likely due to the enormous size and diversity of the naïve TCR repertoire.
Figure 1. TCR landscape of splenic and intra-tumoral CD8 T cells from MC38-bearing mouse.
Intratumoral TCR repertoire is highly reactive to tumor cells in MC38 model
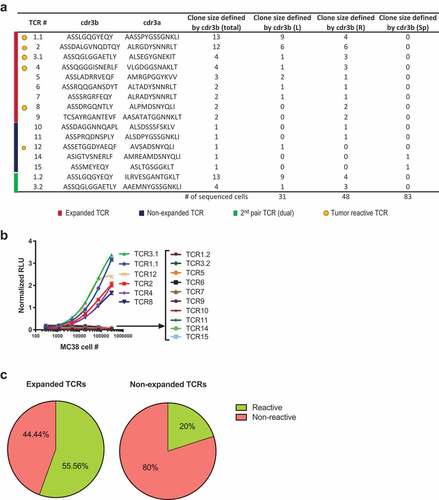
To test whether the TCRs are reactive to MC38 tumor cells, we developed a cell-based luciferase reporter assay using lentiviral transduction of J.RT3-T3.5 cell line (a derivative mutant of Jurkat leukemia cell line, abbreviated as JRT hereafter) for stable surface expression of cloned TCR, as illustrated in Supplementary Figure 1a. The total clone size of each unique TCR is the summary of both tumors and spleen (listed in Supplementary Table 1). We selected all nine expanded TCRs (total clone size ≥ 2) as well as five non-expanded TCRs (total clone size = 1) from different tissues (). The surface expression of human TCR and CD3 was confirmed by FACS (Supplementary Figure 1b). TCR-transduced JRT cells were incubated with a dose-range of in vitro cultured MC38 cells (). The MC38-reactive TCRs were predominantly from expanded TCR clones (5 out of the 6 reactive TCR were from expanded TCR, 83.3%) (). Expanded clones were significantly more reactive to MC38 cells (5 out of 9 expanded clones, 55.6%) in comparison to non-expanded clones (1 out of 5 non-expanded clones, 20%) (). This data suggests that expanded TCRs were more likely to react to the MC38 tumor. In vitro cultured MC38 may not express the same level and breadth of peptide-MHCI repertoire as MC38 tumor cells growing in the tumor microenvironment in vivo. Therefore, it is possible that the non-reactive TCR from the above assay with in vitro cultured MC38 may react to MC38 tumor cells grown under different conditions. To test this, we used IFNγ-treated in vitro cultured MC38 and freshly isolated MC38 cells from tumor tissues as target cells (Supplementary Figure 2) that should express higher levels of MHCI. Both IFNγ-treated and freshly isolated MC38 tumor cells indeed expressed higher levels of MHCI (H2Kb) (Supplementary Figure 2a and C). However, the enhanced expression of MHCI of cultured MC38 or freshly isolated tumor cells did not change the reactivity results of the TCRs (Supplementary Figure 2b and D). These results show that intratumoral TCR repertoire is highly reactive to tumor cells in MC38 model. In addition, the majority of tumor-reactive TCRs are clonally expanded and the expanded TCRs are more likely to be tumor-reactive.
Figure 2. Majority of tumor-reactive TCRs are clonally expanded.
The majority of tumor-reactive TCRs recognize the endogenous retroviral epitope p15E
We next sought to determine the specific antigens that are recognized by these MC38-reactive TCRs. On MC38, we performed Exome-sequencing, RNA-sequencing, and Mass-Spectrometry of eluted MHC-restricted peptides. From Exome and RNA sequencing, we identified expressed somatic mutations, tumor-specific genes, and endogenous retroviral elements. Tumor-specific genes and retroviral elements were derived based on their unique or significantly higher expression comparing to normal mouse tissues. Peptides (8–12 mers) from these mutations, genes, and elements were used for in-silico prediction of binding to MHC alleles H2-Db and H2-Kb. The predicted binder peptides were then used to create search databases for mass-spectrometry data analysis which led to the confirmation of a subset of putative antigenic peptides presented on the cell surface (listed in Supplementary Table 2). We generated a synthetic peptide library based on the confirmed peptides which includes neoantigens, public antigens, and endogenous retroviral antigens, such as p15E peptide previously described in multiple syngeneic tumors including MC38 and B16.Citation14,Citation28
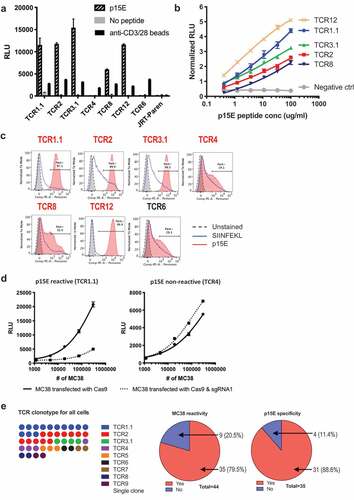
To screen the library, we used TrampC2, a murine prostate cancer cell line in C57BL/6 background, as antigen presentation cells since it does not stimulate any of the MC38-reactive JRT cell lines. We found that 5 out of 6 MC38-reactive TCRs responded specifically to p15E peptide (). All 5 TCRs responded to p15E peptide in a dose-dependent manner (). In addition, 4 of the 5 TCRs that responded to p15E also bound to p15E pentamer as detected by flow cytometry (). Notably, TCR8 demonstrated ambiguous binding to the p15E pentamer although responded to p15E peptide, albeit the lowest response among reactive TCR (-c). This suggests the threshold for pentamer binding to p15E-specific TCR may be higher than that for peptide recognition in the context of MHC expression on cells.
Figure 3. Majority of reactive TILs bear TCRs recognizing a murine endogenous viral epitope.
Previous publications have reported multiple neo-antigens expressed by MC38, with Rpl18 recently shown to be a dominant antigen in this cell line.19,20 Since the majority of TCR identified here were p15E specific and did not recognize any of those antigens, we examined the expression of the published neo-antigens in our cell line and found that not all of them were detected (Supplementary Figure 3a). Specifically, the Rpl18 mutation was not called in our cell line (Supplementary Figure 3b). The different mutation status of MC38 tumor cell lines between laboratories may be due to genetic drift caused by intrinsic genome instability of the tumor cells.Citation29
To verify that p15E peptide is indeed an endogenous epitope, instead of a peptide mimicry, we used CRISPR-Cas9 system to specifically disrupt the coding sequence of this specific epitope (Supplementary Figure 4a). Stimulation of p15E-reactive TCR1.1, but not p15E non-reactive TCR4, was drastically decreased when MC38 were transfected with Cas9 and sgRNA1 (). This result suggested that p15E was indeed the endogenous antigen recognized by p15E reactive TCR1.1 while p15E non-reactive TCR4 recognizes a MC38-specific antigen different from p15E. TCR4 reactivity was slightly increased after p15E disruption, suggesting elimination of p15E made space for presentation of other antigens that could be recognized by TCR4.Citation30 In summary, we found that the majority of MC38-reactive TCRs recognized the endogenous epitope p15E. Indeed, as illustrated in , out of all the clonally expanded cells, the majority of them (79.5%) reacted to MC38, while the majority of all MC38-reactive cells (88.6%) recognized p15E.
TIL reactivity against MC38 mainly comes from a p15E-specific T cell subset
To evaluate if the primary p15E-specific T cells have anti-tumor activity, we isolated endogenous CD8+ TILs from implanted MC38 tumors and tested their reactivity to p15E ex vivo (). We found that the majority (>95%) of p15E-specific CD8+ TILs are PD1+ (p15E+PD1+), while the p15E pentamer negative were ~25% PD-1+ () in agreement with previous findings that PD-1 expression on T cells can correlate with their tumor reactivity.Citation31,Citation32
Figure 4. TIL reactivity against MC38 mainly came from p15E-specific subset.
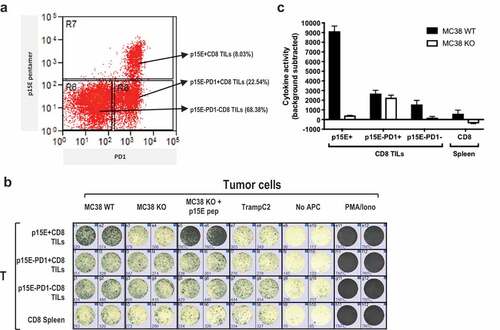
To test the tumor reactivity of these sorted TIL populations, we performed IFNg ELISPOT through incubation with different target cells, including MC38WT, MC38 p15E knockout (MC38KO), MC38KO pulsed with exogenous p15E peptide, as well as irrelevant TrampC2 as negative control. MC38 KO was generated by knocking out p15E coding sequence using CRISPR-Cas9 (Supplementary Figure 4a). The absence of p15E epitope in MC38KO cell line was confirmed by disruption of original genomic DNA and amino acid sequences (Supplementary Figure 4b and S4C) and loss of reactivity of p15E-specific JRT-TCR1.1 in the luciferase assay (Supplementary Figure 4d). Furthermore, p15E-specific CD8+ TILs were absent in MC38KO tumors implanted in vivo (Supplementary Figure 4e). We found that sorted p15E+PD1+ CD8+ TILs were not reactive to MC38KO or TrampC2 cell line (-c). In contrast, the same cells were highly reactive to MC38WT or to MC38 p15E KO cells pulsed with exogenous p15E peptide. The p15E−PD1+CD8+ TILs were tumor reactive in a p15E-independent manner as they responded equally to MC38WT and MC38 p15E KO cells. Reactivity to p15E was barely detected in spleen CD8+ T cells, possibly due to low frequency of circulating tumor-reactive T cells. Interestingly, p15E−PD1−CD8+ T cell population also exhibited some p15E-specific reactivity, suggesting that p15E pentamer might fail to label T cells with low-affinity TCRs, such as TCR8 described in . In summary, the majority of MC38TILs reactivity was primarily due to p15E specificity.
Epigenetic derepression of endogenous retroviral genome drives expression of p15E specifically in tumor cells
p15E (KSPWFTTL) was first described as an immunodominant CTL epitope of B10 B cell lymphoma induced by MCF1223 murine leukemia virus (MuLV).Citation33 It is deemed as an immunodominant epitope in this virus because it is recognized by all CTL clones isolated from the tumor-specific bulk culture. p15E was later identified as a T-cell antigen for many other murine tumors in H-2Kb background, including MC38 and MCA-205.Citation28 These p15E-specific T cells have demonstrated anti-tumor efficacy as cytotoxic T lymphocytes generated in vitro by p15E peptide stimulation of native splenocytes inhibited pulmonary metastasis of MC38, B16, and WP6.Citation28 p15E-reactive TCRs were also identified from B16 TILs.Citation14 p15E-coding AKV proviral genome sequences (J01998.1) can be mapped to C57BL/6 germline genome (Chr 8: 123425804–123434531).Citation34 The germline proviral genome is largely intact except for some point mutations and some deletions in the non-coding region. The genome consists of three viral genes: gag, pol, and env (Supplementary Figure 5a). The initial translation product of env is further cleaved into gp70 (glycoprotein of 70KD) and p15E (envelope protein of 15 KD).
To profile the p15E expression in different tissues, we performed real-time qPCR to measure the mRNA level of env, the open reading frame encoding p15E. We found that the expression of env in MC38 cells and MC38 tumor tissues is about 118,000-fold and 25,500-fold, respectively, higher than normal tissues or TrampC2 cells (). To check whether all 3 genes of the proviral genome were expressed, RNA-seq of tumor cell lines and normal tissues was performed. Consistent with our qPCR results, all three genes were highly expressed in several mouse tumor cell lines of different histological types, but not in normal tissues (). Interestingly, B16F10 only expressed transcripts from gag, but not env or pol, implying a partial inactivation of the proviral genome. Overall, p15E was specifically expressed in certain tumor cells, but not in normal tissues.
Figure 5. p15E-containing AKV proviral genome is actively expressed in tumor cell lines due to epigenetic dysregulation.
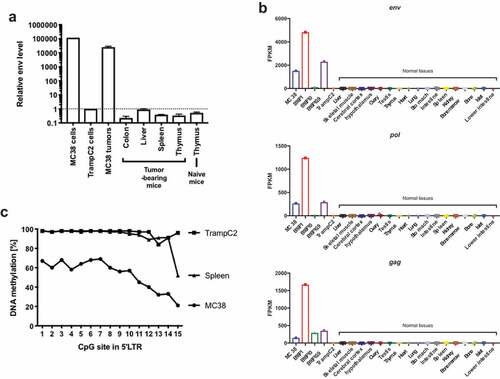
Since p15E transcripts were also detected in other cell lines, including B16F1 and B16F10.9, we tested their abilities to stimulate p15E-specific and non-p15E-specific TCRs identified from MC38. To enhance MHCI expression, the tumor cell lines were pre-treated with IFNγ for 24 hours. As expected, IFNγ pre-treatment enhanced MHCI, MHCII, and PD-L1 expression (Supplementary Figure 6a). To our surprise, although B16F1 and B16F10.9 expressed p15E transcript, they failed to stimulate the p15E-specific TCR (Supplementary Figure 6b). Alignment of the p15E transcripts from MC38 and these B16 derivative cell lines shows that p15E coding sequence was identical and therefore the lack of reactivity was not due to mutations of p15E in the B16 cell lines (Supplementary Figure 6c). To investigate if differences in p15E presentation may be a result of altered antigen processing machinery, we compared the expression of proteasome subunit genes, the key components of the endogenous processing pathway (Supplementary Figure 7). Indeed, we observed multiple proteasome genes were differentially expressed between MC38 and B16 derivatives, such as Psmb8-10, Psmd13, Psmc1, and Psmc3-5. These differences could lead to altered peptide processing and therefore differences in p15E presentation.Citation35
Next, we examined the underlying mechanism of the ectopic expression of p15E in MC38. Failure of epigenetic inhibition may drive expression of endogenous viral gene expression in tumor cells which are known to be genetically unstable.Citation36 To test this, we identified CpG islands in 5ʹ LTR region and performed DNA methylation sequencing (Supplementary Figure 5b). Unlike in TrampC2 and spleen, 5ʹ LTR region in MC38 was highly unmethylated, suggesting the ectopic expression is due to epigenetic derepression (). Our findings showed that although the p15E coding sequence was integrated in C57BL/6 germline genome, its specific expression in certain tumor cells, not in normal tissues, was likely due to an epigenetic derepression.
T cell activation gene expression signature derived from expanded TILs
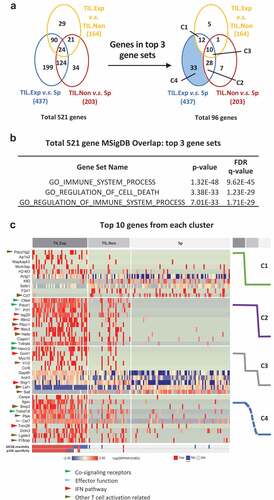
One of the advantages of single-cell sequencing is capturing TCR information and transcriptomic information simultaneously at the single-cell level. Using the TCR clonality information and the tissue of origin, we divided the cells into three groups: expanded TILs (TIL.Exp), non-expanded TILs (TIL.Non Exp.), and spleen T cells (Sp). We performed transcriptome analysis by comparing these three groups, yielding a total of 521 differentially regulated genes which met criteria of fold change larger than 1.5 and p-value smaller than 0.05 (; full list in Supplementary Table 3). TIL.Exp vs Sp group comparison contained the most differentially expressed genes, many of which overlapped with the TIL.Exp vs TIL.Non and TIL.Non vs Sp group comparison. We investigated the enrichment of gene sets in Molecular Signature Database (MSigDB) of the 521 genes and found 96 genes are in the top 3 gene sets that are related to immune system processes and cell death regulation (; complete gene set enrichment results in Supplementary Table 4). Of these 96 genes, 83 (86%) are significantly differentially expressed between TIL.Exp and the Sp cells. We decided to focus on these 83 genes, which fall into 4 categories: TIL.Exp unique signature (C1), tumor tissue signature (C2), gradient signature (C3), and TIL.Exp vs Sp only signature (C4) (). Consistent with previously reported phenotypes of activated T cells, we found that co-signaling receptor genes (Ctla4, Pdcd1, Tnfrsf4, Havcr2, Tnfrsf18 highlighted by green arrows), Citation37 effector function-related genes (Prf1 and Cst7 highlighted by blue arrows), Citation38,Citation39 IFN pathway genes (Isg20, Ifitm2, Ifitm3, Gcnt1, Ifngr1, Trim26 highlighted by red arrows)Citation40 were highly upregulated in expanded TILs compared to non-expanded TILs or splenic CD8+ T cells (). Interestingly, most of these genes were also upregulated in a small portion of non-expanded TILs. One interesting group of genes differentially expressed in expanded TILs labeled by brown arrows consists of a plethora of genes with different functions in T cell activation. For example, Il1r2 was previously shown to be upregulated upon CD8+ T cell activation in vitro.Citation41 The role of Il1 signaling pathway in cancer immunology remains elusive and could be context-dependent, and it deserves further investigation.Citation42 Detailed description and literature references of the genes in are summarized in Supplementary Table 5. Overall, consistently with our previous finding that most tumor reactivity lies in the expanded TIL compartment, the transcriptome analysis suggested that the expanded TILs exhibit an activated phenotype compared with non-expanded TILs or spleen T cells. Limited activation phenotype in non-expanded TILs suggests bystander T cell activation in MC38 tumors. Whether similar gene changes in TIL occur in other tumor types, as well as the functional relevance of these genes require further investigation.
Figure 6. T cell activation gene signature derived from expanded TILs by single-cell RNAseq.
p15E-specific CD8+ T cells showed an activated phenotype and responded to anti-41BB and anti-PD1 treatment
To assess the kinetics of p15E+ CD8+ TILs expansion during MC38 tumor growth, we performed immunophenotyping of MC38 tumors on day 14, 18, and 21 post implantation. Tumor growth over time is illustrated in . Interestingly, the frequency of p15E+ CD8+ T cells remained constant independently of the tumor size or the time post implantation (10–20%) (). Compared to total CD8+ T cells, p15E+ CD8+ T cells exhibit a more activated phenotype with an increased frequency of PD-1+ and CD44+ and decreased frequency of CD62 L+ ( and ).
Figure 7. p15E-specific CD8 T cells showed an activated phenotype and responded to anti-41BB and/or anti-PD1 treatment.
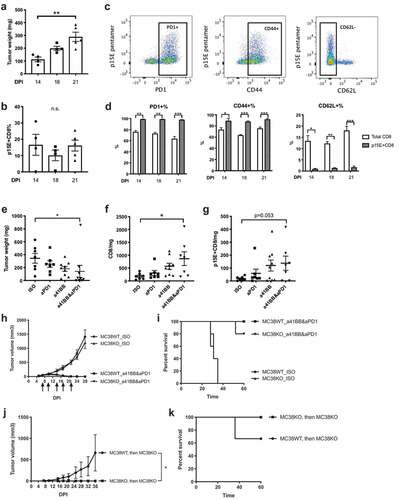
Next, we examined the expansion and the phenotype of p15E+ CD8+ TILs in response to immunotherapy. We treated established MC38 tumors with anti-PD1 alone or anti-41BB alone or a combination treatment. Anti-PD1 and anti-41BB combination therapy synergistically inhibited MC38 tumor growth and significantly increased CD8+ TILs in tumors ( and ). Anti-PD1 and anti-41BB combination treatment also increased p15E+ CD8+ TILs in tumors, although a trend was also observed in monotherapy groups (). In summary, consistent with the single-cell sequencing data shown in , p15E+ CD8+ TILs exhibited an activated phenotype and p15E+CD8+ TILs increased upon combination treatment demonstrating a specific response to immunotherapy.
Removal of p15E epitope impairs the memory response against MC38 tumor
To determine if the primary response and the long-term anti-tumor immunity observed after immunotherapy was dependent on the p15E antigen and the p15E-specific TILs, we used MC38KO cell lines (Supplementary Figure 4). Mice were implanted with MC38WT or MC38KO and treated with either isotype controls or anti-41BB and anti-PD1 combination therapy ( and ). Interestingly, combination treatment induced a robust tumor rejection and increased the survival rate of mice implanted with either MC38WT or MC38KO. This data suggests that while p15E appears to generate a dominant-specific T cell response, it is not required for anti-tumor immune response against MC38. Further, mice that were previously implanted with either MC38WT or MC38KO, treated with combination immunotherapy, and rejected their tumors were re-challenged with MC38KO ( and ). Mice that had previously rejected MC38KO were protected against re-challenge with MC38KO, suggesting that an anti-tumor immune memory was generated against new tumor antigens different from p15E. In contrast, mice that were previously rejected MC38WT had an incomplete protection from MC38KO, suggesting that at least a part of the immune memory against MC38WT was specific for p15E. Further, we also re-challenged the MC38WT- or MC38KO-experienced mice with MC38WT. Unlike rapid tumor progression in naive mice, these mice were fully protected from MC38WT (Supplementary Fig. 8). This suggests that recognition of non-p15E antigens generated in response MC38KO is not diminished in the presence of p15E presented by MC38WT during re-challenge. In conclusion, durable anti-tumor immunity against MC38 generated by combination immunotherapy was partially dependent on p15E, a dominant antigen recognized by T cells. However, in the absence of p15E expression, other immunogenic peptides were sufficient to compensate and stimulate T cells to develop long-term protection.
Discussion
In this study, we used single-cell sequencing to characterize the TCR repertoire of CD8+ T cells in MC38 tumors and spleen. As expected, clonal expansion was observed in MC38 tumors, but not in spleen, implying the active immune response was localized at tumor sites. Although we found highly expanded TCRs from all tumors, MC38 tumors from different mice did not share TCRs. This is likely due to the enormous size of naïve TCR repertoire generated by random V(D)J recombination, resulting in that different TCRs against the same tumor antigens are expanded in different mice. Alternatively, it may be that intra-tumoral TCRs from different mice recognize different tumor antigens, and these unique antigens emerged after implantation and during tumor growth. However, this is unlikely because shared TCRs were found in tumors implanted at different locations on the same mouse. Although “public” TCRs against viral antigens, malignancies, and autoimmunity have been widely described before, Citation43 tumor antigens in MC38 might be less capable of stimulating “public” TCRs. Interestingly, we did not identify shared TCRs in MC38 tumors and spleen from the same mouse, although it has been previously shown that tumor-specific CD8+ T cells can be found in periphery.Citation44 It is likely that tumor-specific T cells in the spleen are present at very low frequencies and below the threshold of detection for the small number of cells we sampled in single-cell sequencing.
Our results suggested that intratumoral TCR repertoire in MC38 tumors was highly reactive. A recent study on human tumors suggested that tumor reactivity of intratumoral TCR repertoire was very variable, with high reactivity in melanoma, but low in high-grade serous ovarian carcinoma and microsatellite stable colorectal adenocarcinoma.Citation12 This highlights that findings on mouse tumor tissues might not warrant clinical translatability. Presumably existing TCR repertoire reactivity against tumors plays a critical role in immune-modulatory therapies; thus, TCR repertoire reactivity should also be considered when selecting mouse tumor models. We also characterized the cellular phenotype of p15E-specific TILs by transcriptome and flow cytometry. p15E-specific TILs enrich an immune activation signature and are PD1+, CD44+, and CD62L−. By comparing expanded TILs, with non-expanded TILs and spleen CD8+, we identified 4 groups of genes based on the statistical significance of the comparisons. Many known genes related to T cell activation were found to be differentially regulated, including co-signaling receptor genes, effector function-related genes, IFN pathway genes, and a group of genes with different functions (detailed description in Supplementary Table 5). Our analysis suggested that T cell phenotypes matched their TCR clonal expansion status. In addition, most of the gene families listed above were also upregulated in a small portion of non-expanded TILs. A potential explanation is that a few expanded cells are “misclassified” as non-expanded TILs due to limited throughput of C1 single-cell sequencing platform. Alternatively, this could be the result of bystander activation of non-expanded TILs in the tumor microenvironment. Limited activation phenotype in non-expanded TILs suggests limited bystander T cell activation in MC38 tumors. This remains to be validated in additional tumor models.
We identified an endogenous retroviral antigen p15E as the immunodominant epitope of MC38. The endogenous viral genome is highly expressed in several tumor cell lines due to epigenetic derepression. Surprisingly, B16F1 and B16F10.9, the derivatives of a murine melanoma cell line highly expressing the endogenous viral genome, were incapable of stimulating p15E-specific JRT reporter cells. Treating them with IFNγ in vitro enhanced MHCI expression but still failed to stimulate p15E-specific reporter cells. Previously published data also suggested that p15E was expressed in B16 parental cell lines and p15E-pulsed DC vaccine or adoptive transfer of p15E-specific T cells inhibited established B16 lung metastasis.Citation45,Citation46 We have shown here that the p15E coding sequence is identical between MC38 and B16 cells. Interestingly, we found differences in the expression of some proteasome genes involved in endogenous processing pathway. It is possible that altered antigen processing machinery results in decreased ability of B16 cell lines to stimulate p15E-specific TCRs, which remains to be confirmed.
Multiple neo-antigens have been reported to be expressed by MC38, including a recently described dominant T cell antigen Rpl18.Citation19,Citation20 The contribution of p15E and these different neo-antigens to the immunogenicity of MC38 are not mutually exclusive. The discovery of unique antigens in MC38 by various groups may be due to different origins and genetic drift of the cell lines over time. As Hos et al. showed, differences in neo-antigen expression were observed between their cell line and the cell line from a commercial source.Citation20 Indeed, we also found differences in expression of these neo-antigens the cell line used in our studies. Since MC38 from different sources are maintained and cultured separately, the genetic drifting might confound the results for neo-antigen discovery in this murine tumor model. A shared depository might be a helpful way to reconcile these results in future studies.
Tumor antigens can be categorized into two groups: tumor-associated self-antigens and tumor-specific antigens. Tumor-associated self-antigens include tissue differentiation antigens and cancer overexpressing antigens, both of which potentially induce thymic or peripheral tolerance. Targeting tumor-associated self-antigens by chimeric antigen receptor-T (CAR-T), antibody-drug conjugates (ADCs), or bispecific antibodies, potentially induce on-target off-tumor toxicity.Citation47 Instead, tumor-specific antigens, including exogenous viral antigens, cancer-testis antigens, and neo-antigens, are considered as better tumor targets due to lack of expression in normal tissues. They may also be good targets for vaccination due to lack of peripheral T cell tolerance to these tumor antigens.Citation21
Here, we discovered that endogenous retroviral antigens strongly contributed to cancer immunogenicity in a murine tumor model. Despite their genome integrations, lack of expression in normal tissues puts endogenous retroviral antigens into tumor-specific category. Human endogenous retroviruses (HERVs) comprise about 8% of human genome, majority of which are epigenetically silenced or contain mutations preventing their expression.Citation48 Although there are several examples that HERVs drive malignancies at DNA- and protein-levels, most of tumor-associated HERV overexpression is considered as a “passenger” effect due to a general epigenetic dysregulation.Citation49 HERV-K expression has been detected in melanoma, breast cancer, ovarian cancer, lymphoma, germ cell cancer, and prostate cancer tissues.Citation50–Citation58 Furthermore, antibodies and CTLs against HERV-K were also detected in patients.Citation50–Citation58 Antibodies, ADCs, and CAR-Ts targeting HERV-K envelope proteins demonstrated tumor-specific killing in preclinical models of multiple human tumor cell lines demonstrating the potential of HERV-K targeted therapies.Citation50,Citation52 Further, epigenetic drugs, such as DNA methyltransferase inhibitors and histone demethylase inhibitors, enhance endogenous retroviral gene expression, which could be combined with endogenous retroviral targeting therapeutics.Citation59–Citation61 Overall, endogenous retroviral antigens represent a promising class of tumor targets for future drug development.
In summary, our results revealed an endogenous retroviral antigen as the immunodominant epitope of MC38, which provides a useful tool to study tumor-specific immune response (Supplementary Fig. 9). Additional antigen characterization of different murine tumor models could shed light on model selection for pre-clinical drug testing. These studies highlight that the sources and levels of immunogenicity are important metrics to consider when choosing animal models to faithfully predict clinical results. Finally, endogenous retroviral antigens represent a novel category of tumor antigens which could be readily targeted in clinical settings.
Competing interests
All authors are employees of Regeneron Pharmaceuticals, Inc.
Supplemental Material
Download ()Supplementary Material:
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19(3):133–16. doi:10.1038/s41568-019-0116-x.
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi:10.1038/nature12477.
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. doi:10.1038/nature13954.
- Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, Kuo F, Kendall SM, Requena D, Riaz N, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359(6375):582–587. doi:10.1126/science.aao4572.
- Schatz DG, Ji Y. Recombination centres and the orchestration of V(D)J recombination. Nat Rev Immunol. 2011;11(4):251–263. doi:10.1038/nri2941.
- De Simone M, Rossetti G, Pagani M. Single Cell T Cell Receptor Sequencing: techniques and Future Challenges. Front Immunol. 2018;9:1638. doi:10.3389/fimmu.2018.01638.
- Han A, Glanville J, Hansmann L, Davis MM. Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat Biotechnol. 2014;32(7):684–692. doi:10.1038/nbt.2938.
- Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell. 2017;169(7):1342–56e16. doi:10.1016/j.cell.2017.05.035.
- Robert L, Tsoi J, Wang X, Emerson R, Homet B, Chodon T, Mok S, Huang RR, Cochran AJ, Comin-Anduix B, et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin Cancer Res. 2014;20(9):2424–2432. doi:10.1158/1078-0432.CCR-13-2648.
- Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14(2):135–146. doi:10.1038/nrc3670.
- Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8(4):299–308. doi:10.1038/nrc2355.
- Scheper W, Kelderman S, Fanchi LF, Linnemann C, Bendle G, MAJ DR, Hirt C, Mezzadra R, Slagter M, Dijkstra K, et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat Med. 2019;25(1):89–94. doi:10.1038/s41591-018-0266-5.
- Ahmadzadeh M, Pasetto A, Jia L, Deniger DC, Stevanovic S, Robbins PF, Rosenberg SA. Tumor-infiltrating human CD4+ regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci Immunol. 2019;4(31). doi:10.1126/sciimmunol.aao4310.
- Shitaoka K, Hamana H, Kishi H, Hayakawa Y, Kobayashi E, Sukegawa K, Piao X, Lyu F, Nagata T, Sugiyama D, et al. Identification of tumoricidal TCRs from tumor-infiltrating lymphocytes by single-cell analysis. Cancer Immunol Res. 2018;6(4):378–388. doi:10.1158/2326-6066.CIR-17-0489.
- van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, Knuth A, Boon T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254(5038):1643–1647. doi:10.1126/science.1840703.
- Sahin U, Tureci O, Schmitt H, Cochlovius B, Johannes T, Schmits R, Stenner F, Luo G, Schobert I, Pfreundschuh M, et al. Human neoplasms elicit multiple specific immune responses in the autologous host. Proc Natl Acad Sci U S A. 1995;92(25):11810–11813. doi:10.1073/pnas.92.25.11810.
- Cox AL, Skipper J, Chen Y, Henderson RA, Darrow TL, Shabanowitz J, Engelhard V, Hunt D, Slingluff C. Identification of a peptide recognized by five melanoma-specific human cytotoxic T cell lines. Science. 1994;264(5159):716–719. doi:10.1126/science.7513441.
- Gee MH, Han A, Lofgren SM, Beausang JF, Mendoza JL, Birnbaum ME, Bethune MT, Fischer S, Yang X, Gomez-Eerland R, et al. Antigen identification for orphan T cell receptors expressed on tumor-infiltrating lymphocytes. Cell. 2018;172(3):549–63e16. doi:10.1016/j.cell.2017.11.043.
- Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515(7528):572–576. doi:10.1038/nature14001.
- Hos BJ, Camps MGM, Jvd B, Tondini E, Ende T, Ruano D, Franken K, Janssen GMC, Ru AHD, Filippov DV, et al. Identification of a neo-epitope dominating endogenous CD8 T cell responses to MC-38 colorectal cancer. OncoImmunology. 2019;1673125. doi:10.1080/2162402x.2019.1673125.
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–221. doi:10.1038/nature22991.
- Glanville J, Huang H, Nau A, Hatton O, Wagar LE, Rubelt F, Ji X, Han A, Krams SM, Pettus C, et al. Identifying specificity groups in the T cell receptor repertoire. Nature. 2017;547(7661):94–98. doi:10.1038/nature22976.
- Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellstrom KE, Mittler RS, Chen L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med. 1997;3(6):682–685. doi:10.1038/nm0697-682.
- Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410(6832):1107–1111. doi:10.1038/35074122.
- Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65(3):1089–1096.
- Bai Y, Wang D, Li W, Huang Y, Ye X, Waite J, Barry T, Edelmann KH, Levenkova N, Guo C, et al. Evaluation of the capacities of mouse TCR profiling from short read RNA-seq data. PLoS One. 2018;13(11):e0207020. doi:10.1371/journal.pone.0207020.
- Wang B, Zhang W, Jankovic V, Golubov J, Poon P, Oswald EM, Gurer C, Wei J, Ramos I, Wu Q, et al. Combination cancer immunotherapy targeting PD-1 and GITR can rescue CD8+T cell dysfunction and maintain memory phenotype. Sci Immunol. 2018;3(29). doi:10.1126/sciimmunol.aat7061.
- Yang JC, Perry-Lalley D. The envelope protein of an endogenous murine retrovirus is a tumor-associated T-cell antigen for multiple murine tumors. J Immunother. 2000;23(2):177–183. doi:10.1097/00002371-200003000-00001.
- Homet Moreno B, Zaretsky JM, Garcia-Diaz A, Tsoi J, Parisi G, Robert L, Meeth K, Ndoye A, Bosenberg M, Weeraratna AT, et al. Response to programmed cell death-1 blockade in a murine melanoma syngeneic model requires costimulation, CD4, and CD8 T cells. Cancer Immunol Res. 2016;4(10):845–857. doi:10.1158/2326-6066.CIR-16-0060.
- Boulanger DSM, Eccleston RC, Phillips A, Coveney PV, Elliott T, Dalchau N. A mechanistic model for predicting cell surface presentation of competing peptides by MHC class i molecules. Front Immunol. 2018;9:1538. doi:10.3389/fimmu.2018.01538.
- Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124(5):2246–2259. doi:10.1172/JCI73639.
- Fernandez-Poma SM, Salas-Benito D, Lozano T, Casares N, Riezu-Boj JI, Mancheno U, Elizalde E, Alignani D, Zubeldia N, Otano I, et al. Expansion of tumor-infiltrating CD8(+)T cells expressing PD-1 improves the efficacy of adoptive T-cell therapy. Cancer Res. 2017;77(13):3672–3684. doi:10.1158/0008-5472.CAN-17-0236.
- Sijts AJ, Ossendorp F, Mengede EA, van den Elsen PJ, Melief CJ. Immunodominant mink cell focus-inducing murine leukemia virus (MuLV)-encoded CTL epitope, identified by its MHC class I-binding motif, explains MuLV-type specificity of MCF-directed cytotoxic T lymphocytes. J Immunol. 1994;152:106–116.
- Herr W. Nucleotide sequence of AKV murine leukemia virus. J Virol. 1984;49(2):471–478. doi:10.1128/JVI.49.2.471-478.1984.
- Sijts EJ, Kloetzel PM. The role of the proteasome in the generation of MHC class I ligands and immune responses. Cell Mol Life Sci. 2011;68(9):1491–1502. doi:10.1007/s00018-011-0657-y.
- Kassiotis G. Endogenous retroviruses and the development of cancer. J Immunol. 2014;192(4):1343–1349. doi:10.4049/jimmunol.1302972.
- Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13(4):227–242. doi:10.1038/nri3405.
- Lichtenheld MG, Olsen KJ, Lu P, Lowrey DM, Hameed A, Hengartner H, Podack ER. Structure and function of human perforin. Nature. 1988;335(6189):448–451. doi:10.1038/335448a0.
- Perisic Nanut M, Sabotic J, Jewett A, Kos J. Cysteine cathepsins as regulators of the cytotoxicity of NK and T cells. Front Immunol. 2014;5:616. doi:10.3389/fimmu.2014.00616.
- Bailey CC, Zhong G, Huang IC, Farzan M. IFITM-family proteins: the cell’s first line of antiviral defense. Annu Rev Virol. 2014;1:261–283. doi:10.1146/annurev-virology-031413-085537.
- Wang M, Windgassen D, Papoutsakis ET. Comparative analysis of transcriptional profiling of CD3+, CD4+ and CD8+ T cells identifies novel immune response players in T-cell activation. BMC Genomics. 2008;9:225. doi:10.1186/1471-2164-9-225.
- Mantovani A, Barajon I, Garlanda C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol Rev. 2018;281(1):57–61. doi:10.1111/imr.12614.
- Li H, Ye C, Ji G, Han J. Determinants of public T cell responses. Cell Res. 2012;22(1):33–42. doi:10.1038/cr.2012.1.
- Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, Prickett TD, Gartner JJ, Crystal JS, Roberts IM, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. 2016;22(4):433–438. doi:10.1038/nm.4051.
- Zeh HJ 3rd, Perry-Lalley D, Dudley ME, Rosenberg SA, Yang JC. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J Immunol. 1999;162:989–994.
- Kershaw MH, Hsu C, Mondesire W, Parker LL, Wang G, Overwijk WW, Lapointe R, Yang JC, Wang RF, Restifo NP, et al. Immunization against endogenous retroviral tumor-associated antigens. Cancer Res. 2001;61(21):7920–7924.
- Ilyas S, Yang JC. Landscape of tumor antigens in T cell immunotherapy. J Immunol. 2015;195(11):5117–5122. doi:10.4049/jimmunol.1501657.
- Grandi N, Tramontano E. HERV envelope proteins: physiological role and pathogenic potential in cancer and autoimmunity. Front Microbiol. 2018;9:462. doi:10.3389/fmicb.2018.00462.
- Bannert N, Hofmann H, Block A, Hohn O. HERVs new role in cancer: from accused perpetrators to cheerful protectors. Front Microbiol. 2018;9:178. doi:10.3389/fmicb.2018.00178.
- Krishnamurthy J, Rabinovich BA, Mi T, Switzer KC, Olivares S, Maiti SN, Plummer JB, Singh H, Kumaresan PR, Huls HM, et al. Genetic engineering of T cells to target HERV-K, an ancient retrovirus on melanoma. Clin Cancer Res. 2015;21(14):3241–3251. doi:10.1158/1078-0432.CCR-14-3197.
- Wang-Johanning F, Radvanyi L, Rycaj K, Plummer JB, Yan P, Sastry KJ, Piyathilake CJ, Hunt KK, Johanning GL. Human endogenous retrovirus K triggers an antigen-specific immune response in breast cancer patients. Cancer Res. 2008;68(14):5869–5877. doi:10.1158/0008-5472.CAN-07-6838.
- Wang-Johanning F, Rycaj K, Plummer JB, Li M, Yin B, Frerich K, Garza JG, Shen J, Lin K, Yan P, et al. Immunotherapeutic potential of anti-human endogenous retrovirus-K envelope protein antibodies in targeting breast tumors. J Natl Cancer Inst. 2012;104(3):189–210. doi:10.1093/jnci/djr540.
- Johanning GL, Malouf GG, Zheng X, Esteva FJ, Weinstein JN, Wang-Johanning F, Su X. Expression of human endogenous retrovirus-K is strongly associated with the basal-like breast cancer phenotype. Sci Rep. 2017;7(1):41960. doi:10.1038/srep41960.
- Wang-Johanning F, Liu J, Rycaj K, Huang M, Tsai K, Rosen DG, Chen D-T, Lu DW, Barnhart KF, Johanning GL, et al. Expression of multiple human endogenous retrovirus surface envelope proteins in ovarian cancer. Int J Cancer. 2007;120(1):81–90. doi:10.1002/ijc.22256.
- Rycaj K, Plummer JB, Yin B, Li M, Garza J, Radvanyi L, Ramondetta LM, Lin K, Johanning GL, Tang DG, et al. Cytotoxicity of human endogenous retrovirus K-specific T cells toward autologous ovarian cancer cells. Clin Cancer Res. 2015;21(2):471–483. doi:10.1158/1078-0432.CCR-14-0388.
- Contreras-Galindo R, Kaplan MH, Leer P, Verjat T, Ferlenghi I, Bagnoli F, Giusti F, Dosik MH, Hayes DF, Gitlin SD, et al. Human endogenous retrovirus K (HML-2) elements in the plasma of people with lymphoma and breast cancer. J Virol. 2008;82(19):9329–9336. doi:10.1128/JVI.00646-08.
- Kleiman A, Senyuta N, Tryakin A, Sauter M, Karseladze A, Tjulandin S, Gurtsevitch V, Mueller-Lantzsch N. HERV-K(HML-2) GAG/ENV antibodies as indicator for therapy effect in patients with germ cell tumors. Int J Cancer. 2004;110(3):459–461. doi:10.1002/ijc.11649.
- Agoni L, Guha C, Lenz J. Detection of human endogenous retrovirus K (HERV-K) transcripts in human prostate cancer cell lines. Front Oncol. 2013;3:180. doi:10.3389/fonc.2013.00180.
- Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, Li Y, Chen H, Yang H, Hsu P-H, et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell. 2018;174(3):549–63 e19. doi:10.1016/j.cell.2018.05.052.
- Liu M, Zhang L, Li H, Hinoue T, Zhou W, Ohtani H, El‐Khoueiry A, Daniels J, O’Connell C, Dorff TB, et al. Integrative epigenetic analysis reveals therapeutic targets to the DNA methyltransferase inhibitor guadecitabine (SGI-110) in hepatocellular carcinoma. Hepatology. 2018;68(4):1412–1428. doi:10.1002/hep.30091.
- Liu M, Thomas SL, DeWitt AK, Zhou W, Madaj ZB, Ohtani H, Baylin SB, Liang G, Jones PA. Dual inhibition of DNA and histone methyltransferases increases viral mimicry in ovarian cancer cells. Cancer Res. 2018;78(20):5754–5766. doi:10.1158/0008-5472.CAN-17-3953.