
ABSTRACT
Antitumor immunity is mediated by Th1 CD4+ and CD8+ T lymphocytes, which induce tumor-specific cytolysis, whereas Th17 CD4+ T cells have been described to promote tumor growth. Here, we explored the influence of IL-17 on the ability of therapeutic vaccines to induce the rejection of tumors in mice using several adjuvants known to elicit either Th1 or Th17-type immunity. Immunization of mice with Th1-adjuvanted vaccine induced high levels of IFN-γ-producing T cells, whereas injection with Th17-promoting adjuvants triggered the stimulation of both IL-17 and IFN-γ-producing T cells. However, despite their capacity to induce strong Th1 responses, these Th17-promoting adjuvants failed to induce the eradication of tumors. In addition, the systemic administration of IL-17A strongly decreases the therapeutic effect of Th1-adjuvanted vaccines in two different tumor models. This suppressive effect correlated with the capacity of systemically delivered IL-17A to inhibit the induction of CD8+ T-cell responses. The suppressive effect of IL-17A on the induction of CD8+ T-cell responses was abolished in mice depleted of neutrophils, clearly demonstrating the role played by these cells in the inhibitory effect of IL-17A in the induction of antitumor responses. These results demonstrate that even though strong Th1-type responses favor tumor control, the simultaneous activation of Th17 cells may redirect or curtail tumor-specific immunity through a mechanism involving neutrophils. This study establishes that IL-17 plays a detrimental role in the development of an effective antitumor T cell response and thus could strongly affect the efficiency of immunotherapy through the inhibition of CTL responses.
Introduction
The correlation found for many human tumors between the infiltration of CD8+ T cells and the prolonged survival of patientsCitation1 has led to the development of therapeutic cancer vaccines based on the induction of effective antitumor CD8+ T-cell responses. In addition, several studies have established that patients with high expression of the Th1 cluster in colorectal cancer show prolonged disease-free survival, in contrast to the poor diagnosis of patients with high expression of the Th17 cluster.Citation2 A higher proportion of intratumoral IL-17-producing cells also correlates with poor survival in hepatocellular carcinoma patients.Citation3
Th17 CD4+ T cells produce IL-17A, IL-17F, IL-21, and IL-22 and contribute to host defenses against certain infections, particularly in mucosal tissues.Citation4 They also participate in the pathogenesis of various allergic, autoimmune, and inflammatory diseases and play a role in transplantation.Citation5,Citation6 IL-17 is a central player in immunity and is produced by several cell types other than Th17 cells, such as γδ T and natural killer cells, as well as by innate lymphoid cells 3 (ILC3).Citation7
Many studies have demonstrated that IL-17 can promote tumor growth via its effects on vascular endothelium and increased neovascularization. Indeed, the transfection of IL-17 into human tumor cell lines increases their growth in nude and SCID miceCitation8,Citation9 by promoting angiogenesis and tumor resistance to antiangiogenic therapy.Citation10 IL-17 was shown to induce tumor development through the increased expression of Stat3 and infiltration of myeloid cells into the tumor in an experimental model of carcinogen-induced skin cancer.Citation11 In accordance with these findings, blockade of IL-17A at the tumor site using an adenovirus vector expressing siRNA against the mouse IL-17A gene suppressed tumor growth and induced the activation of cytotoxic T-cell responses.Citation12 Similar results were obtained in IL-17RA deficient mice in which increased infiltration of the tumor by CD8+ T cells was also shown.Citation13
However, other studies have shown that Th17 cells can confer protective antitumor immunity.Citation14 Indeed, in contrast to the results obtained in immunodeficient mice, the growth of IL-17-producing tumors was significantly inhibited in immunocompetent mice relative to that of mock-transfected tumors.Citation15–Citation17 In addition, tumor growth and lung metastasis were enhanced in IL-17 deficient mice, suggesting that endogenous IL-17 plays a protective role in tumor immunity.Citation18,Citation19 The adoptive transfer of tumor-specific Th17 cells promoted the activation of cytotoxic T cells against the tumorCitation19 and induced the eradication of large established melanomas.Citation20
Overall, these data provide supportive evidence that IL-17 may play a dual role in human tumor immunity and that the activation of tumor-specific Th17 cells could promote the therapeutic efficacy of cancer vaccines.
Here, we explored the influence of IL-17 on the ability of therapeutic vaccines to induce the rejection of tumors in mice using several adjuvants known to elicit either Th1 or Th17-type immunity. Immunization of mice with ovalbumin (OVA) and CpG formulated in DOTAP, a TLR9 agonist and a Th1-promoting adjuvant, induced high levels of IFN-γ-producing T cells, whereas injection of OVA with Th17-promoting adjuvants, such as the β-1,3-glucan curdlan from the cell wall of fungi,Citation21 the cationic polysaccharide chitosan,Citation22 or the mycobacterial cell-wall component trehalose-6, 6ʹ-dimycolate (TDM),Citation23 triggered the stimulation of both IL-17 and IFN-γ-producing T cells. However, despite their capacity to induce strong Th1 responses, these Th17-promoting adjuvants failed to induce the eradication of OVA-expressing tumors. In addition, we demonstrate that systemic administration of recombinant IL-17A strongly decreases the therapeutic effect of CpG-adjuvanted vaccines in two different tumor models. This suppressive effect correlated with the capacity of systemically delivered IL-17A to inhibit the induction of antigen–specific CD8+ T-cell responses by the therapeutic vaccines. The suppressive effect of IL-17A on the induction of CD8+ T-cell responses was abolished in mice depleted of neutrophils, clearly demonstrating the role played by these cells in the inhibitory effect of IL-17A in the induction of antitumor responses.
These results demonstrate that even though strong Th1-type responses favor tumor control, the simultaneous activation of Th17 cells may redirect or curtail tumor-specific immunity through a mechanism involving neutrophils. This study establishes that IL-17 plays a detrimental role in the development of an effective antitumor T cell response and thus could strongly affect the efficiency of immunotherapy through the inhibition of CTL responses.
Materials and methods
Mice
Six to eight-week-old, female, pathogen-free C57BL/6J mice were purchased from Charles River Laboratories. IL-17RA deficient miceCitation24 and Rag-/- OT-I T-cell receptor transgenic mice, specific for the Kb-restricted OVA257-264 (SIINFEKL) epitope, and IFNAR- and IFNγ- deficient mice were provided by the animal facilities of the Pasteur Institute. All mouse strains used in this study were on a C57BL/6J background. Studies involving mice were validated by CETEA ethics committee number 0068 (Institut Pasteur, Paris, France) and the French Ministry of Research (MESR 00672.02).
Reagents
The DMT adjuvant contained dimethyl dioctadecyl ammonium bromide (DDA) (250 μg/mouse), monophosphoryl lipid A (MPL) (25 μg/mouse), and trehalose dicorynomycolate (TDM) (25 μg/mouse) (Sigma-Aldrich Chimie). Curdlan (Dectin-1 agonist) was purchased from Wako (Osaka, Japan) and 2 mg/mouse was used. Chitosan was obtained from Novamatrix (Sandvika, Norway) and 1 mg/mouse was used. Detoxified CyaA of Bordetella pertussis, carrying a truncated form of the E7 protein from HPV-16 (CyaA-E7), was prepared as previously described.Citation25 CpG-B 1826 (5-TTCCATGACGTTCCTGACGTT-3) was synthesized by Sigma and 30 µg mixed with 60 μg N-[1-(2,3-dioleoyloxyl)propyl]-NNNtrimethylammoniummethyl sulfate (DOTAP, Roche) in 100 μL PBS (Gibco, ThermoFisher) was used per mouse.
Tumor experiments
TC-1 tumor cellsCitation26 expressing the HPV-16 E6 and E7 proteins were obtained from the American Type Culture Collection (LGC Promochem) and grown in complete medium consisting of RPMI 1640 supplemented with GlutaMAX (Gibco, ThermoFisher), 10% FCS (Gibco, ThermoFisher), antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin; Gibco, ThermoFisher), and 2 × 10−5 M 2-mercaptoethanol (Gibco, ThermoFisher), 0.4 mg/mL geneticin G418 (Gibco, ThermoFisher), and 0.2 mg/mL hygromycin B (Roche). B16-OVA tumor cells were kindly provided by L. Rosthein and L. Sigal (University of Massachusetts, Worcester, MA) and cultured in complete medium supplemented with 2 mg/mL geneticin G418 (Gibco, ThermoFisher) and 60 mg/mL hygromycin B (Roche).
B16-OVA and TC-1 cells were injected s.c. (2.5.105 and 5.105 cells, respectively) into C57BL/6 mice or IFN-γ-, IFNAR-, or IL-17RA- deficient mice. For B16-OVA experiments, mice received PBS or adjuvant alone or 100 µg OVA alone or with DMT, CpG-B and DOTAP, or curdlan. For TC-1 experiments, mice were immunized i.v. with CyaA-E7 (50 μg/mouse) together with CpG-B and DOTAP (30 μg and 60 μg/mouse, respectively). In some experiments, mice also received i.p. injections of 1 or 3 μg murine IL-17A protein (R&D systems; Bio-Techne). Following tumor inoculation, tumor growth was assessed every two to three days by measuring the size of the tumor with a digital caliper. Tumor size is presented as the average of two perpendicular diameters (millimeters). Mice were sacrificed when the tumor diameter reached 20 mm.
Cell isolation
Spleens were harvested, treated for 20 min with 400 U/mL collagenase type IV and 50 µg/mL DNase I (Boehringer Mannheim), and mechanically disrupted and filtered to obtain single-cell suspensions. The tumors were harvested, incubated for 45 min with 400 U/mL collagenase D and 50 μg/mL DNase I, and dissociated using the gentleMACS dissociator (Miltenyi Biotec, Paris, France, program mimpTumor-01-01). The dissociated tumors were then filtered to obtain single-cell suspensions and resuspended in PBS supplemented with 1% FCS.
Analysis of CTL responses by in vivo killing, ELISPOT assays, and SIINFEKL Dextramers
C57BL/6 J mice were i.p. injected with PBS or 1 or 3 µg IL-17A in 200 µL PBS daily from day −1 to day 7. On day 0, they received s.c. injections of 30 µg CpG-B, 60 µg DOTAP and 100 µg OVA protein at the base of the tail. On day 7, splenocytes from naive C57BL/6J mice were loaded with 1 ng/mL SIINFEKL OVA peptide or left unloaded for 30 min at 37°C. Then, OVA peptide-loaded splenocytes were labeled for 15 min in the dark at room temperature with 2.5 µM CFSE and unloaded splenocytes with 0.25 µM CFSE. Cells were then mixed in a 1:1 ratio before i.v. injection (107 total cells in 200 µL PBS per mouse). On day 8, mice were euthanized, the cells collected for image acquisition, and the CFSEhi and CFSElo populations analyzed. The percentage of cytotoxicity was calculated as (1-percentage of CFSEhi/percentage of CFSElo) normalized to the ratio in control mice. The percentage of SIINFEKL/H-2Kb dextramer+ CD8+ T cells was also assessed by FACS, whereas OVA-specific IFNγ-producing splenocytes were detected by ELISPOT.
Adoptive transfer of CD8+ T cells
Splenic OT-I CD8+ T cells were purified by magnetic negative selection using the CD8+ T-cell isolation kit from Miltenyi Biotec following the manufacturer’s procedure. Then, the cells were labeled with 10 µM CFSE (Molecular Probes) for 15 min at room temperature and 5.105 cells injected into mice by the i.v. route. Mice were immunized i.v. one day later with OVA and CpG-B and DOTAP and also received daily i.p. injections of PBS or IL-17A for four days. The CD8+ T-cell response was analyzed five days after immunization.
Depletion of neutrophils
IL-17A-treated or untreated C57BL/6J mice were i.v. injected one day before immunization and 1, 3, and 5 days after immunization with 200 µg anti-Ly6G (BioXcell; clone 1A8) or the control isotype (Rat IgG2a, κ, anti-trinitrophenol Ab, clone 2A3).
Flow cytometry analysis
Cell suspensions were incubated with rat anti-CD16/32 mAb (clone 2.4G2) to block binding to Fc receptors before staining with diverse fluorescent dye-conjugated mAbs. The monoclonal antibodies (mAbs) used for staining were the following: APC-conjugated anti-CD3 (clone 17A2), anti-CD11c (clone HL3), anti-CD25 (clone 7D4), anti-CD40L (clone MR1), and anti-Ki67 (clone SolA15); APC-eF780-conjugated anti-CD45 (clone 104); PB conjugated anti-CD3 (clone 17A2), anti-CD4 (clone RM4-5), anti-CD11b (clone M1/70), anti-CD44 (clone IM7), anti-CD73 (clone eBioTY/11.8), and anti-Ly6C (clone HK1.4); PerCP- and PerCP-Cy5.5-conjugated 7AAD, anti-B220 (clone RA3-6B2), anti-CD4 (clone RM4-5), and anti-CD49b (clone DX5); PE-Cy7-conjugated anti-CD11b (clone M1/70), anti-CD11c (clone N418), anti-CD19 (clone 1D3), anti-CD28 (clone 37.51), anti-CD39 (clone 24DMS1), anti-CD62L (clone MEL-14), and anti-NK1.1 (clone PK136); PE-conjugated Annexin V, anti-CD8 (clone 53–6.7), anti-CD69 (clone H1.2F3), anti-FasL (clone MFL3), anti-ICOS (clone 7E.17G9), anti-Ly6G (clone 1A8-Ly6g), anti-PD-1 (clone J43), anti-PD-L1 (clone MIH5), and anti-IL-17RA (clone PAJ-17R); FITC-conjugated anti-CD8 (clone 53–6.7), anti-CD25 (clone 7D4), anti-CD27 (clone LG.7F9), anti-CD40 (clone HM40-3), anti-FoxP3 (clone FJK-16S), and anti-Ly6G (clone 1A8); EF650-conjugated anti-CD11b (clone M1/70); BUV395-conjugated anti-CD45 (clone OX-1); and biotin-conjugated anti-CD49b (clone DX5), anti-Fas (clone 15A7), anti-ICOSL (clone HK5.3), and anti-PD-L1 (clone MIH5). All mAbs were purchased from either BD Biosciences, Biolegend, or eBioscience (ThermoFisher). Streptavidin-PE-conjugated H-2Kb/SIINFEKL Dextramer was purchased from Immudex. Streptavidin-PE-conjugated H-2Db/E749-57 tetramers were purchased from MBL International Corporation.
FoxP3+ and Ki67+ cells were detected by intracellular staining with FITC-conjugated anti-FoxP3 and APC-conjugated anti-Ki67 mAbs, respectively, after treatment with Cytofix/Cytoperm, following the manufacturer’s protocol (eBioscience, ThermoFisher). Annexin V+ 7AAD− and Annexin V+ 7AAD+ apoptotic cells and Annexin V− 7AAD+ necrotic cells were detected by staining with PE-conjugated Annexin V and PerCP-conjugated 7AAD following the manufacturer’s protocol (BD Pharmingen). The cells were acquired using a LSR Fortessa (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR).
Statistical analysis
Student’s t-test, the Mann-Whitney test, and Kaplan-Meier survival analyses were performed to calculate the statistical significance of differences between groups using Prism software (GraphPad Software, Inc.). P values < .05 were considered statistically significant.
Results
The curdlan and DMT adjuvants are less efficient than CpG in delaying the growth of B16-OVA expressing melanoma
We assessed the role of IL-17 in the activation of therapeutic anti-cancer immune responses by comparing the protection achieved against the growth of B16-OVA tumor cells by immunization of mice with OVA and CpG-B complexed with DOTAP, a Th1 adjuvant, or DMT or curdlan, two adjuvants that elicit mixed Th1/Th17 responses. DMT is composed of dimethyl dioctadecyl ammonium bromide (DDA), monophosphoryl lipid A (MPL), and synthetic trehalose dicorynomycolate (TDM). The Th1/Th17 adjuvanticity of DMTCitation27 is controlled by the macrophage-inducible Ca2+-dependent lectin receptor (Mincle), whereas curdlan is a selective Dectin-1 agonist.Citation28
We first compared the immune responses of mice immunized with OVA alone or with CpG-B, curdlan, or DMT as adjuvant. As expected, spleen cells from mice immunized with OVA and CpG-B produced only IFN-γ after in vitro stimulation with OVA, whereas the splenocytes of mice that received OVA with either DMT or curdlan produced both IFN-γ and IL-17A (Figure S1A). Both IL-1β and IL-6 were produced by mice injected with CpG-B, curdlan, or DMT, whereas the production of IFN-α was triggered by both DMT and CpG-B but not curdlan. In contrast, only CpG-B induced the production of IL-12p40 (Figure S1B).
We determined whether the production of IL-17 can influence the induction of therapeutic immune responses by grafting C57BL/6 mice with B16-OVA tumor cells, followed by immunization 5, 13, and 21 days later with OVA alone or OVA with DMT, curdlan, or CpG-B and monitoring of tumor progression (). Immunization with OVA and CpG-B resulted in strong protection against tumor growth, with 46% of the treated mice rejecting the tumor. In contrast, only a low level (21-25%) of protection was obtained in mice immunized with OVA and DMT or curdlan and no protection following administration of OVA or adjuvant alone.
Figure 1. The curdlan and DMT adjuvants are less efficient than CpG in delaying the growth of B16-OVA-expressing melanoma. C57BL6/J (a-c), C57BL6/J and IFN-γ-/- (d-e), and C57BL6/J and IFNAR-/- (f-g) mice were injected s.c. with 2.5.105 B16-OVA cells and subsequently injected s.c. with PBS, DMT, curdlan or CpG-B alone or with 100 μg OVA 5, 13, and 21 days later. (h-i) C57BL6/J mice were injected s.c. with 2.5.105 B16-OVA cells and subsequently injected s.c. one or two times (2x) with 100 μg OVA alone or with DMT or CpG-B 5, 13, and 21 days later. Control mice received only PBS. (a) Each curve represents the tumor diameter of an individual mouse. The results represent the cumulative data of three independent experiments (n = 8 mice per group). (b, d, f, h) The results represent the percentage of tumor-free mice 80 days after the injection of B16-OVA cells. The numbers of mice that rejected the tumor from the total number of mice are indicated for each group. (c, e, g, i) Kaplan-Meier plot of mouse survival. ns: not significant, *p < .05, ***p < .001, as determined by the logrank test. (i) The p value between OVA+CpG (2x) and OVA+CpG//OVA+DMT = 0.0266 whereas the p value between OVA+CpG and OVA+CpG (2x) = 0.695. (b-i) The results represent the cumulative data of two experiments.
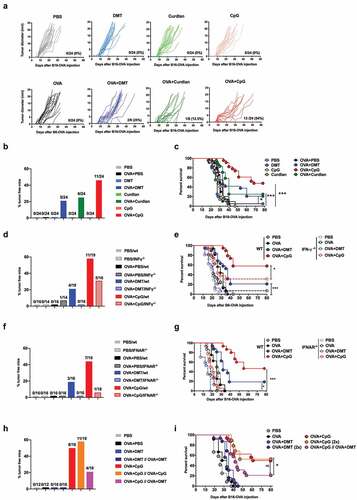
We determined whether type I and II interferons were involved in the control of tumor growth induced by immunization with OVA and the adjuvants by similarly treating IFN-γ () or IFNAR () deficient mice grafted with B16-OVA tumor cells. Immunization with OVA and CpG or DMT induced a lower level of protection in these mice than in C57BL/6 mice. Thus, both IFN-γ and type I IFN played a crucial role in tumor protection, regardless of the adjuvant used.
We then analyzed whether Th17 responses could affect the tumor protection induced by immunization with OVA/CpG-B. Mice were grafted with B16-OVA tumor cells and then received one or two injections of OVA/CpG-B, OVA/DMT or a combination of the two (-). As expected, strong protection against tumor growth was obtained in mice immunized with one or two injections of OVA/CpG-B, whereas no protection was obtained when DMT was used as adjuvant. In addition, mice immunized with a combination of OVA/CpG-B and OVA/DMT showed significantly less protection than those immunized with OVA/CpG-B alone, suggesting that the immune responses induced by DMT played a detrimental role in the CpG-induced protective antitumor immunity.
The depletion of CD8+ T cells in mice grafted with B16-OVA tumor cells and then treated with OVA/CpG-B or OVA/DMT fully abrogated the tumor protection induced by immunization. In contrast, the depletion of CD4+ T cells did not significantly affect the therapeutic activity of the OVA/CpG-B or OVA/DMT vaccine, although the percentage of surviving mice increased (Figure S2). These results demonstrate that the protection induced by OVA/CpG-B against tumor growth was mediated by CD8+ T cells, suggesting that mixed Th1/Th17 promoting adjuvants could affect the induction of tumor-specific CD8+ T cells.
We confirmed these results using chitosan, a cationic polysaccharidic adjuvant, which induces Th1/Th17 responses.Citation22 Chitosan promoted the induction of OVA-specific Th1/Th17 responses when combined with CpG-B, as demonstrated by the production of both IFN-γ and IL-17A by OVA-stimulated splenocytes (Figure S3A), confirming previous results.Citation22 Simultaneous administration of chitosan with OVA/CpG-B fully abolished the tumor protection induced by OVA/CpG-B alone (Figure S3B-C). These results confirm that Th17 responses strongly inhibited the tumor-specific therapeutic response induced by the Th1 adjuvant.
Overall, these results demonstrate that a Th17 environment strongly affects the tumor protection mediated by CD8+ T-cell responses.
IL-17A strongly inhibits the therapeutic efficacy of antitumoral vaccines
We determined whether IL-17 was involved in the decreased inhibition of tumor growth when Th1/Th17 adjuvants were used for immunization by grafting mice with B16-OVA tumor cells following by immunization 5, 13 and, 21 days later with OVA/CpG-B and treatment every three days with 1 µg recombinant IL-17A or PBS from day 5 to day 32. Although OVA/CpG-B immunization strongly reduced tumor growth, as expected, this therapeutic activity was markedly reduced in mice treated with IL-17A ().
Figure 2. IL-17A decreases the therapeutic efficacy of CpG-adjuvanted OVA and CyaA-E7 vaccines. (a-b) C57BL6/J mice were injected s.c. with 2.5.105 B16-OVA cells and subsequently injected s.c. with PBS or 100 μg OVA alone or with CpG-B 5, 13, and 21 days later. Mice received i.p. administration of PBS or 1 μg IL-17A five days after grafting of the tumor cells and every three days thereafter until day 32. (c-d) C57BL6/J mice were injected s.c. with 5.105 TC-1 cells and injected i.v. with PBS or 50 μg CyaA-E7 and CpG-B 20 days later. Mice received i.p. administration of PBS or 1 μg IL-17A 20 days after grafting of the tumor cells and every three days thereafter until day 42. (e-f) C57BL6/J and IL-17RA deficient mice were injected s.c. with 2.5.105 B16-OVA cells and subsequently injected s.c. with 100 μg OVA alone or with DMT or CpG-B 5, 13, and 21 days later. Some OVA/CpG immunized mice also received i.p injections of either PBS or 1 μg of IL-17A five days after grafting of the tumor cells and every three days thereafter until day 32. (a, c, e) Each curve represents the tumor diameter of an individual mouse. The results represent the cumulative data from three independent experiments (n = 8 mice per group). The number and percentage of mice that rejected the tumor from the total number of mice are indicated. (b, d, f) Kaplan-Meier plot of mouse survival. ns: not significant, *p < .05, **p < .01, ***p < .001, as determined by the logrank test.
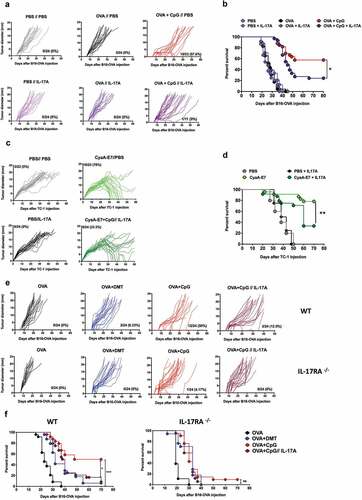
We confirmed these results in a second tumor model. C57BL/6 mice were grafted with TC-1 tumor cells derived from primary lung epithelial cells, expressing the E6 and E7 oncoproteins of HPV-16,Citation26 and immunized with the CyaA-E7 vaccine, a detoxified form of the adenylate cyclase of B. pertussis carrying a truncated form of the E7 protein of HPV-16.Citation25 This vector was previously demonstrated to induce strong specific CTL and Th1 responses.Citation29 Indeed, a single immunization of TC-1-bearing mice with CyaA-E7 and CpG-B at day 20 strongly reduced tumor growth, with 78% of the mice rejecting the tumor (). Administration of IL-17A to these mice greatly diminished the therapeutic efficacy of the CyaA-E7 vaccine, thus confirming the detrimental effect of IL-17A on the induction of protective antitumor immune responses.
We then analyzed the effect of IL-17A administration on OVA/CpG-B induced antitumor immunity using IL-17A receptor (IL-17RA) deficient mice. However, B16-OVA cells grew faster in IL-17RA deficient mice than in wildtype mice, confirming previous studies using IL-17A deficient mice,Citation19 and no therapeutic effect was observed following OVA/CpG-B immunization ().
IL-17A inhibits the activation of specific CD8+ T-cell responses
We next grafted C57BL/6 mice with TC-1 tumor cells, followed by immunization at day 20 with CyaA-E7 and treatment or not with IL-17A, to decipher the mechanisms by which IL-17A reduced the therapeutic activity of the CyaA-E7 vaccine. At day 30, 91% of the mice immunized with CyaA-E7/CpG-B had rejected their tumors, whereas only 61% of the mice that had received CyaA-E7/CpG-B and IL-17A were able to control the growth of the TC-1 tumor cells (Figure S4A). We then analyzed the leucocyte infiltration into the spleen and tumors of mice that rejected their tumors (R: regressor mice) and those that were unable to control tumor progression (P: progressor mice) at day 31 (Figure S4B-C). The number of CD8+ T cells was significantly higher in the tumors of mice immunized with CyaA-E7/CpG-B, whereas the number of CD8+ T cells was lower in progressor but not regressor mice that received IL-17A (Figure S4B-C).
We then assessed the anti-E7 CD8+ T-cell response in the spleen and tumor by flow cytometry staining with the E749-57H-2Db tetramer. We observed a high number of CD8+ T cells in TC-1-bearing mice immunized with CyaA-E7, both in the spleen and tumor (Figure S4D). In the tumor, almost all CD8+ T cells were specific for the E749-57 peptide. In mice treated with IL-17A, these specific CD8+ T-cell responses were significantly lower in the spleens and tumors of progressor mice, whereas regressor mice showed no significant difference.
These results suggest that IL-17A reduced the efficacy of CpG-adjuvanted OVA or CyaA-E7 vaccine by inhibiting the activation of tumor-specific CD8+ T cells. We then analyzed the CD8+ T-cell responses of C57BL/6 mice immunized with OVA/CpG-B and treated daily either with PBS or 1 µg recombinant IL-17A for one week after immunization to confirm this hypothesis.
IL-17A treated mice showed significantly less OVA-specific in vivo killing and fewer IFN-γ ELISPOT responses, as well as a smaller number of OVA257-264-specific T cells, than PBS-treated mice ( and ). The inhibitory effect of IL-17A on the induction of CD8+ T cells was dose dependent and we detected a lower number of OVA-specific CD8+ T cells and IFN-γ producing cells in the spleens of mice treated with a higher dose (3 µg) of recombinant IL-17A (). In addition, the inhibitory effect of IL-17A was dependent on its interaction with the IL-17A receptor, as it did not occur in IL-17RA deficient mice ().
Figure 3. IL-17A inhibits the induction of CTL responses by OVA/CpG. C57BL6/J (a) and IL-17RA deficient (b) mice were immunized s.c. with PBS or 100 µg OVA and CpG-B. They also received i.p. injections of PBS or 1 μg IL-17A one day before immunization and then every day for one week. (c-d) C57BL6/J (WT) and IFN-γ-/- (c) or IFNAR-/- mice (d) were immunized s.c. with PBS or 100 µg OVA and CpG-B. (a-d) Seven days after immunization, the anti-OVA CD8+ T cell response was assessed by an in vivo killing assay (IVK), IFN-γ ELISPOT (IFN-γ) and SIINFEKL/H-2Kb dextramer staining (MHC Dextramer). The results are expressed as the percentage of OVA-specific lysis for CTL activity; IFN-γ spot-forming cells (SFC) per 106 splenocytes for ELISPOT and the percentage of SIINFEKL/H-2Kb dextramer+ among total CD8+ splenocytes for dextramer staining. The results represent the cumulative data from two to six independent experiments and each dot represents an individual mouse. The mean ± SEM is shown for each group. ns: non-significant, *p < .05, **p < .01, ***p < .001, ****p < .0001, as determined by the unpaired Student’s t-test.
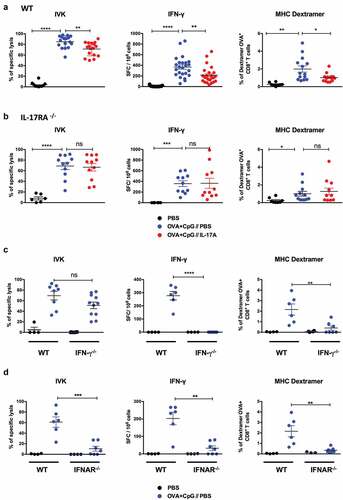
Figure 4. Absence of a direct effect of IL-17A on the CD8+ T-cell responses induced by OVA/CpG. (a) C57BL6/J mice were immunized i.v. with PBS or 100 µg OVA and CpG-B. They received i.p. injections of PBS or IL-17A (1 μg or 3 µg) one day before immunization and then every day for one week. Seven days after immunization, the anti-OVA CD8+ T-cell response was assessed by SIINFEKL/H-2Kb dextramer staining (MHC Dextramer) and IFN-γ ELISPOT (IFN-γ). The results are expressed as the percentage of SIINFEKL/H-2Kb dextramer+ among total CD8+ splenocytes for dextramer staining and IFN-γ spot-forming cells (SFC) per 106 splenocytes for ELISPOT. (b) C57BL6/J and IL-17RA deficient recipient mice were injected with CFSE-labeled (10 µM) OT-1 cells (5.105) isolated from the spleens of OT-1 Rag1KO transgenic mice (95% purity). Twenty-four hours later, recipient mice received a single i.v. injection of PBS or OVA and CpG-B. They received i.p. injections of PBS or IL-17A (3 µg) one day before immunization and then every day for four days. Mice were euthanized at day 5 after immunization and the anti-OVA CD8+ T cell response assessed by SIINFEKL/H-2Kb dextramer staining (MHC Dextramer) and IFN-γ ELISPOT (IFN-γ). Results are representative of two independent experiments and each dot represents an individual mouse. The mean ± SEM is shown for each group. ns: non-significant, *p < .05, **p < .01, as determined by the unpaired Student’s t-test.
The CTL response induced by the Th1 adjuvanted-vaccine was also dependent on both IFN-γ and type I IFN signaling, as CD8+ T-cell responses induced by OVA/CpG-B were much smaller in IFN-γ and IFNAR deficient than wildtype mice (). These results correlated with the absence of tumor protection observed in these immunodeficient mice following vaccination ().
Thus, overall, these results clearly demonstrate that IL-17A strongly affects the therapeutic efficacy of antitumoral vaccines by inhibiting the activation of specific CD8+ T-cell responses.
IL-17A indirectly inhibits the induction of the CD8+ T-cell response
We next sought to decipher the mechanisms by which IL-17A reduces the efficacy of the therapeutic activity of antitumoral vaccines. We first determined whether this cytokine can directly affect the growth of TC-1 and B16-OVA tumor cells. Although these tumor cells express the IL-17 receptor A (Figure S5A), IL-17A did not modify their proliferation (Figure S5B).
The IL-17 receptor is expressed by mouse B cells, T cells (CD8+, CD4+ T cells and Treg cells), dendritic cells, and neutrophils (Figure S5C-D). We thus tested whether IL-17A can directly activate CD4+ or CD8+ T cells. However, we did not observe any modification of their calcium flux (Figure S6A) or their expression of activation markers (Figure S6B) following in vitro incubation of these cells with various concentrations of IL-17A. In contrast, IL-17A significantly inhibited the proliferation of both CD4+ and CD8+ T cells from C57BL/6 mice following in vitro polyclonal activation by anti-CD3 antibodies, without affecting the responses of IL-17RA deficient T cells (Figure S6C). This inhibition correlated with the induction of T-cell apoptosis by IL-17A and T cells from C57BL/6 mice also showed significantly greater cell death than those from IL-17RA deficient mice following anti-CD3 stimulation (Figure S6D-E).
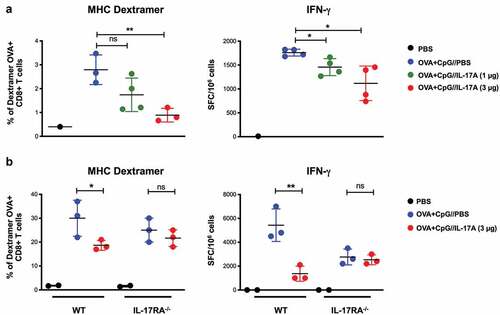
These results suggest that IL-17A may directly affect activation of the CD8+ T-cell response by inhibiting T-cell proliferation and inducing T-cell death. Thus, to determine if IL-17A directly affect the activation of CD8+ T cell responses, we transferred OVA-specific transgenic OT-I T cells, which express the IL-17 receptor, to either C57BL/6 or IL-17RA deficient mice, which were then immunized with OVA/CpG-B and treated daily with PBS or 3 µg recombinant IL-17A for one week.
The OVA-specific T-cell response was greatly reduced when OT-I cells were transferred to C57BL/6 mice treated with IL-17A (), as expected. In contrast, there was no significant difference after the transfer of OT-I cells to IL-17RA deficient mice, showing that the observed reduction of the CD8+ T-cell response was not due to a direct effect of IL-17A on OVA-specific CD8+ OT-I T cells but required the expression of the IL-17 receptor by the recipient mice.
IL-17A induces the recruitment of neutrophils to the spleen
We next analyzed the effect of daily injections of IL-17A on hematopoietic cells in the spleen, bone marrow, and thymus of C57BL/6 mice versus those of IL-17RA deficient mice to identify the cell population(s) required for the suppressive effect of this cytokine. The spleens of IL-17A treated mice showed a significantly higher number of cells, in parallel with a lower number of bone marrow cells (). These effects required IL-17 signaling, as they were not observed in IL-17RA deficient mice. Despite this increase in cell number, the distribution of B and T lymphocytes and dendritic cells was not affected in the spleens of IL-17A treated mice (-).
Figure 5. Systemic administration of IL-17A leads to the recruitment of neutrophils. C57BL/6J and IL-17RA deficient mice were i.p. injected daily with either PBS or 1 µg IL-17A for one week and cell populations characterized by flow cytometry at day 7. (a) Number of total live CD45+ hematopoietic cells in the spleen, bone marrow, and thymus. (b) Percentage of CD19+ B220+ B cells, CD3+ T cells, and CD11c+ cDCs in the spleens of C57BL/6J and IL-17RA deficient mice. (c) Percentage of CD4+ and CD8+ T cells among total CD3+ T cells (right panel) and of FoxP3+ (Treg) and FoxP3− (Teff) among CD4+ T cells (left panel) in the spleens of C57BL/6J and IL-17RA deficient mice. Results are expressed as the mean ± SEM (n = 3 mice per group) and represent the cumulative results of three independent experiments. (d) C57BL/6J mice were injected i.p. with 1 or 3 µg IL-17A or PBS. The percentage of neutrophils among total live CD45+ cells in the spleens of the mice was determined by flow cytometry 3, 6, or 24 h later, according to their expression of Ly6C and Ly6G (Ly6C+, Ly6G+) as gated in Supplementary Figure S7A. (e) Total number of neutrophils (Ly6C+ Ly6G+ cells) in the spleens of C57BL/6J and IL-17RA deficient mice injected i.p. with 1 or 3 µg IL-17A or PBS for seven days. Results are expressed as the mean ± SEM (n = 3 mice per group) and are representative of two independent experiments. ns: non-significant, *p < .05, **p < .01, ***p < .001, ****p < .0001, as determined by the unpaired Student’s t-test.
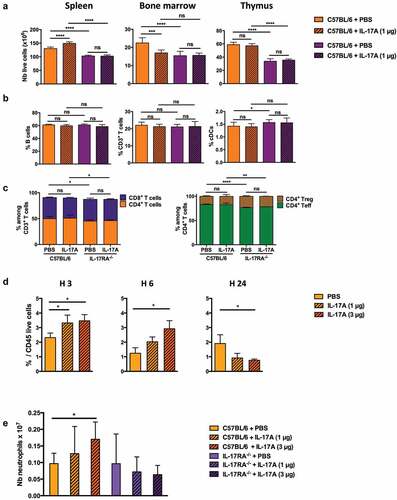
We then studied the recruitment of myeloid cells to the spleens of mice treated with either 1 or 3 µg injections of IL-17A. We analyzed CD11b+ cells, including macrophages, conventional DCs, and neutrophils, for their expression of Ly6C and Ly6G at various times after treatment (Figure S7A). The percentage of neutrophils greatly increased in the spleens of treated mice three and six hours after IL-17A administration, whereas the other CD11b+ populations were not significantly affected, with the exception of the CD11b+ Ly6Chigh subset (monocytes 1), which showed a transient increase three hours after IL-17A injection ( and Figure S7B). The spleens of mice treated for one week with 3 µg IL-17A also showed an increase in the number of neutrophils (). Such neutrophil recruitment was dependent on IL-17A signaling, as it did not occur in IL-17RA deficient mice.
The suppressive effect of IL-17A on the induction of the CD8+ T-cell response is mediated by neutrophils
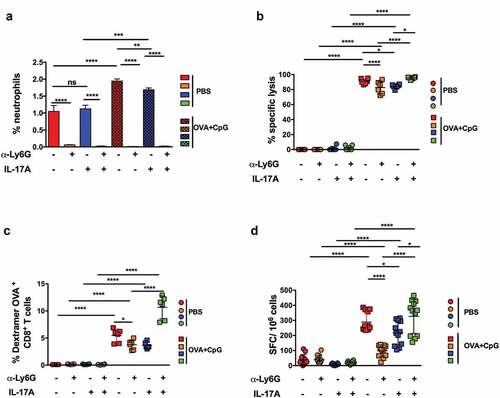
We treated mice immunized with OVA/CpG-B with anti-Ly6G antibodies to determine whether neutrophils were involved in the inhibition of the CD8+ T-cell response by IL-17A (). Such treatment fully depleted the neutrophil population but also affected the distribution of several CD11b+ cell subsets, such as Ly6Chi and Ly6C+ Ly6Glo cells, which were much more abundant in mice receiving anti-Ly6G antibodies (Figure S8).
Figure 6. The suppressive effect of IL-17 on CD8+ T-cell responses is mediated by neutrophils. C57BL6/J mice were immunized s.c. with PBS or 100 µg OVA and CpG-B at day 0. Mice were treated with anti-Ly6G mAbs or control isotype one day before immunization and every two days for one week. They also received PBS or 1 μg IL-17A by the i.p. route one day before immunization and every day for one week. Seven days later, the percentage of splenic CD11b+ CD11c− Ly6C+ Ly6 G+ neutrophils among total CD45+ live cells was determined by flow cytometry (a) and the anti-OVA CD8+ T-cell response assessed by an in vivo killing assay (b), SIINFEKL/H-2Kb dextramer staining (c), and IFN-γ ELISPOT (d). The results are expressed as the percentage of OVA-specific lysis for CTL activity, IFN-γ spot-forming cells (SFC) per 106 splenocytes for ELISPOT, and the percentage of SIINFEKL/H-2Kb dextramer+ among total CD8+ splenocytes for dextramer staining. The data represent the cumulative results of two independent experiments and each dot represents an individual mouse. The mean ± SEM is shown for each group. Results are expressed as the mean ± SEM and represent the cumulative data of two independent experiments. ns: non-significant, *p < .05, **p < .01, ***p < .001, ****p < .0001, as determined by the unpaired Student’s t-test.
The treatment of mice immunized with OVA/CpG-B by anti-Ly6G antibodies also induced an increase in the total number of splenocytes, including CD4+ and CD8+ T cells, which was not observed in mice treated with IL-17A. However, such treatment did not modify the percentage of CD3+ T cells or the CD4+/CD8+ T-cell ratio among total splenic cells (Figure S9).
Finally, we analyzed the effect of neutrophil depletion on the CTL responses of mice immunized with OVA/CpG-B. As previously observed, treatment with anti-Ly6G antibodies fully depleted the neutrophil population in mice immunized or not with OVA/CpG-B and treated with either PBS or IL-17A ().
The depletion of neutrophils from mice immunized with OVA/CpG-B and treated only with PBS induced a significant decrease in CTL activity, measured by in vivo killing, and the number of OVA-specific CD8+ T cells, analyzed either by dextramer or ELISPOT (-). In contrast, OVA-specific CD8+ T-cell responses were much higher following the depletion of neutrophils from mice immunized with OVA/CpG-B and treated with 1 µg IL-17A. We obtained similar results in mice treated with 3 µg IL-17A and anti-Ly6G antibodies (Figure S10). In the absence of neutrophils, the OVA-specific CD8+ T-cell responses induced in mice immunized with OVA/CpG-B and treated with IL-17A were even higher than the responses obtained following vaccination by OVA/CpG-B alone.
Overall, these results reveal the crucial role played by IL-17A and neutrophils in the induction of antitumor immune responses.
Discussion
Here, we show that immunization with Th17-promoting adjuvants, such as DMT or curdlan, fail to induce the rejection of tumors, in contrast to a Th1-promoting adjuvant. In addition, the systemic administration of recombinant IL-17A strongly inhibited the therapeutic effect of CpG-adjuvanted vaccines, as well as the induction of antigen-specific CD8+ T-cell responses. This suppressive effect of IL-17A was abolished in mice depleted of neutrophils, clearly demonstrating the role of these cells in the inhibition of antitumor immune responses by IL-17A.
OVA adjuvanted with the bacterial cell-wall components TDM and curdlan promoted mixed Th1/Th17 responses but failed to reproduce the therapeutic potency of a Th1-promoting CpG/DOTAP adjuvanted vaccine. The therapeutic activity of the OVA/CpG-B vaccination was mediated by CD8+ T cells and strongly dependent upon both type I IFN and IFN-γ. Both DMT and curdlan induced high levels of IFN-γ, comparable to that produced after immunization with CpG, confirming previous studies,Citation22,Citation23 and DMT also induced the production of type I IFN. Thus, the lack of therapeutic activity of Th17-promoting adjuvants was probably not due to the lack of production of these cytokines. In addition, our observation of significantly less protection of mice immunized with a combination of OVA/CpG-B and OVA/DMT than those immunized with OVA/CpG-B alone strongly suggests that the immune responses induced by Th17-promoting adjuvants inhibited Th1-induced protective antitumor immunity.
We confirmed this hypothesis by demonstrating that the chronic administration of IL-17A strongly inhibits the therapeutic effect of CpG-adjuvanted vaccines in two different tumor models. The inhibitory effect of IL-17A was dependent upon IL-17A receptor signaling, as it did not occur in IL-17RA deficient mice. These results are in direct contrast to previous studies supporting a protective role for IL-17 in anti-tumor host defenses. However, we also showed that B16-OVA grows faster in IL-17RA receptor deficient than in wildtype mice, supporting the promotion of tumor immunity by IL-17. These results are in accordance with previous studies showing that IL-17 deficient mice are more susceptible to the growth of the poorly immunogenic B16-F10 melanomaCitation19 or MC38 murine colon adenocarcinoma.Citation18 Here, we also showed the absence of a therapeutic effect of OVA/CpG-B in IL-17RA receptor deficient mice, despite the fact that these mice developed CTL responses similar to those of wildtype mice after OVA/CpG-B immunization. IL-17 deficient mice did not present major differences in their distribution of splenic B or T lymphocytes, although we observed significantly lower numbers of cells in the spleen, bone marrow, and thymus than those of control mice. Thus, our results support the conclusion that a constitutive deficiency in IL-17 signaling may indeed promote the tumor growth by a mechanism which is yet to be elucidated and may be independent of the induction of innate or adaptive immune responses. However, better control of tumor growth was observed in an IL-17A deficient mice in a murine model of hepatocellular carcinoma,Citation30 supporting the conclusion that the IL-17 pro or antitumor effect is related to cancer type.Citation31
The present study clearly establishes the detrimental role of IL-17 in the induction of adaptive antitumor immunity in wildtype mice. Indeed, the administration of IL-17A strongly inhibited the induction of CD8+-specific CTL responses. In contrast to our results, the transfer of Th17 cells have been claimed to promote the activation of anti-tumor cytotoxic T cellsCitation19 and reduce tumor growth in several prevention and therapeutic models. However, in the aforementioned study, Th17 cells were obtained by in vitro polarization of CD4+ T cells from OT-II transgenic mice. Although the study concluded that transferred Th17 cells maintained their Th17 phenotype and did not convert to Th1 cells, this hypothesis cannot be totally excluded. Indeed, in vitro polarization of transgenic T cells specific for the tyrosinase-related protein (TRP) showed TRP-specific Th17 cells to eradicate established melanoma more efficiently than Th1 cells.Citation20 However, tumor rejection induced by such Th17 cells was completely inhibited by anti-IFN-γ antibodies, but not neutralizing antibodies against IL-17A or IL-13, suggesting that tumor rejection was mediated by Th17 cells that converted to Th1 cells. Conversion of in vitro polarized Th17 cells into IFN-γ producing Th1 cells has indeed been shown in NOD SCID mice.Citation32 Human Th17 cells specific for a tumor antigen have also been shown to progressively convert to IFN-γ-secreting cells as they differentiate into effector T cells in vivo.Citation33 In addition, Th17 cells can also transdifferentiate into regulatory T cells, contributing to the resolution of inflammation, showing their instability and plasticity.Citation34
Our study shows that IL-17A can significantly inhibit the proliferation of T cells in vitro following anti-CD3 stimulation, in correlation with its capacity to induce T-cell apoptosis. However, the inhibitory effect of IL-17 was not due to a direct effect on T cells, but mainly mediated by the recruitment of neutrophils. Indeed, we observed no suppression of CTL induction when OT-I T cells were transferred to IL-17RA deficient mice, in contrast to the inhibition observed after transfer to wildtype mice.
We demonstrated the involvement of neutrophils in the capacity of IL-17A to inhibit the activation of CD8+ T-cell responses by the depletion of neutrophils following treatment with the 1A8 anti-Ly6G specific monoclonal antibodies, which have been shown to deplete neutrophils but preserve Gr-1-expressing blood monocytes.Citation35 The depletion of neutrophils from mice immunized with OVA/CpG-B significantly reduced the OVA CD8+ T-cell response, whereas after IL-17A treatment, it strongly increased this response. This response was even significantly higher than that obtained following vaccination by OVA/CpG-B in the presence of neutrophils. These results suggest that neutrophils recruited following IL-17A administration may exert a different effect than at steady state. Indeed, the heterogeneity of neutrophils has been well documented in diseases and cancer.Citation36 The capacity of IL-17A to trigger granulopoiesis in mice, leading to neutrophil expansion and recruitment, is also well established.Citation37 In addition, IL-17 has been shown to promote the accumulation of polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) in human colorectal cancer.Citation38 PMN-MDSCs are defined in mice as CD11b+Ly6G+Ly6Clo Citation39 whereas we defined neutrophils as CD11b+Ly6G+Ly6C+ in this study. Both populations share the same origin and many phenotypic features.Citation40 Whether PMN-MDSCs represent a subset of neutrophils or are a distinct granulocyte population is still a subject of debate. Our study suggests that neutrophils recruited by the chronic administration of IL-17A possess immunosuppressive properties, mimicking the inhibitory activity of PMN-MDSCs. Such an interaction between IL-17 produced by γδ T cells and neutrophils, leading to the inhibition of cytotoxic T-cell responses, has been demonstrated in mice bearing mammary tumors.Citation41 In contrast, neutrophils could promote the induction of CD8+ T-cell responses in the absence of IL-17A treatment through their capacity to present antigen.Citation42
In addition to the relevance of our study for the development of cancer vaccines that promote efficient rejection of tumors, our results may also explain the role of the microbiota in immunotherapy. Indeed, the human microbiota has been shown to affect the response to immunotherapy.Citation43 Many human cancers are located in mucosal sites and mucosal imprinting has been shown to be crucial for the capacity of vaccine-induced CD8+ T cells to inhibit the growth of such tumors.Citation44 In addition, it is well established that the composition of the gut microbiota affects the composition of T-cell subsets in the gut. In particular, segmented filamentous bacteria (SFB) are sufficient to induce the differentiation of Th17 cells, which subsequently produce IL-17 and IL-22.Citation45 SFB antigens presented by intestinal dendritic cells drive the differentiation of mucosal Th17 cells.Citation46 In addition, Th17 cells do not represent the only source of IL-17. γδ T cells and a variety of immune cells, including invariant NK cells and innate ILC3 cells, also produce IL-17.Citation47 Thus, the gut microbiota could favor the induction of Th17 cells against tumor antigens or the production of IL-17, leading to the inhibition of CD8+ T-cell responses. Our results therefore suggest that the microbiota could strongly limit the development of CTL responses through the stimulation of IL-17, thus interfering with the efficacy of immunotherapy. However, SFB induces the recruitment of neutrophils through the stimulation of IL-17A production, which in turn limits the expansion of SFB. Indeed, neutrophil depletion in mice results in significantly augmented SFB levels and Th17 responses.Citation48 Thus, the administration of monoclonal antibodies against either IL-17A or its receptor could represent a potential strategy to enhance the efficacy of immunotherapy. Indeed, local targeting of IL-17A in experimental lung adenocarcinoma has been shown to result in a reduction of tumor load.Citation49 Importantly, IL-17A has been shown to promote PDL1 expression in human and mouse tumorsCitation50 and the treatment of mice with combined anti-IL-17A and anti-PDL1 antibodies induced significant antitumor effects in a murine model of breast cancer. Several monoclonal antibodies targeting the IL-17/IL17RA axis have successfully passed phase III clinical trials and have been approved for the treatment of psoriasis. Such monoclonal antibodies could represent important tools for combined immunotherapy with immune checkpoint inhibitors to improve antitumor immunity.
Declaration of interest statement
The authors declare no competing financial or no financial interests.
Author contributions
C.F, M.O., F.R., P.R., A.T., and E.A.T. designed and performed the experiments, analyzed the results, and provided valuable input concerning the manuscript. I.C. provided the IL-17RA deficient mice. G.D designed and performed the experiments, analyzed the results, and wrote the manuscript. C.L. conceived and supervised the study, designed the experiments, analyzed and discussed the data, and wrote the manuscript.
Supplemental Material
Download ()Acknowledgments
We gratefully acknowledge Marilyne Davi and Daniel Ladant for providing the CyaA-E7. This work was supported by the Ligue Nationale Contre le Cancer (Equipe Labellisée 2017).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–14. doi:10.1126/science.1129139.
- Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, Berger A, Bruneval P, Fridman W-H, Pagès F, et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011;71(4):1263–1271. doi:10.1158/0008-5472.CAN-10-2907.
- Zhang JP, Yan J, Xu J, Pang XH, Chen MS, Li L, Wu C, Li S-P, Zheng L. Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J Hepatol. 2009;50:980–989. doi:10.1016/j.jhep.2008.12.033.
- Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi:10.1084/jem.20081463.
- Abadja F, Sarraj B, Ansari MJ. Significance of T helper 17 immunity in transplantation. Curr Opin Organ Transplant. 2012;17:8–14. doi:10.1097/MOT.0b013e32834ef4e4.
- Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, et al. Differential roles of interleukin-17A and −17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30(1):108–119. doi:10.1016/j.immuni.2008.11.009.
- Veldhoen M. Th17 cells require you to chew before you swallow. Immunity. 2017;46:8–10. doi:10.1016/j.immuni.2016.12.016.
- Tartour E, Fossiez F, Joyeux I, Galinha A, Gey A, Claret E, Sastre-Garau X, Couturier J, Mosseri V, Vives V, et al. Interleukin 17, a T-cell-derived cytokine, promotes tumorigenicity of human cervical tumors in nude mice. Cancer Res. 1999;59:3698–3704.
- Numasaki M, Fukushi J, Ono M, Narula SK, Zavodny PJ, Kudo T, Robbins PD, Tahara H, Lotze MT. Interleukin-17 promotes angiogenesis and tumor growth. Blood. 2003;101:2620–2627. doi:10.1182/blood-2002-05-1461.
- Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, Vernes J-M, Jiang Z, Meng YG, Peale FV, et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med. 2013;19:1114–1123. doi:10.1038/nm.3291.
- Wang L, Yi T, Zhang W, Pardoll DM, Yu H. IL-17 enhances tumor development in carcinogen-induced skin cancer. Cancer Res. 2010;70(24):10112–10120. doi:10.1158/0008-5472.CAN-10-0775.
- Hayata K, Iwahashi M, Ojima T, Katsuda M, Iida T, Nakamori M, Ueda K, Nakamura M, Miyazawa M, Tsuji T, et al. Inhibition of IL-17A in tumor microenvironment augments cytotoxicity of tumor-infiltrating lymphocytes in tumor-bearing mice. PLoS One. 2013;8(1):e53131. doi:10.1371/journal.pone.0053131.
- He D, Li H, Yusuf N, Elmets CA, Li J, Mountz JD, Xu H. IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells. J Immunol. 2010;184(5):2281–2288. doi:10.4049/jimmunol.0902574.
- Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol. 2010;10:248–256. doi:10.1038/nri2742.
- Benchetrit F, Ciree A, Vives V, Warnier G, Gey A, Sautes-Fridman C, Fossiez F, Haicheur N, Fridman WH, Tartour E. Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood. 2002;99:2114–2121. doi:10.1182/blood.V99.6.2114.
- Hirahara N, Nio Y, Sasaki S, Minari Y, Takamura M, Iguchi C, Dong M, Yamasawa K, Tamura K. Inoculation of human interleukin-17 gene-transfected Meth-A fibrosarcoma cells induces T cell-dependent tumor-specific immunity in mice. Oncology. 2001;61(1):79–89. doi:10.1159/000055357.
- Gnerlich JL, Mitchem JB, Weir JS, Sankpal NV, Kashiwagi H, Belt BA, Porembka MR, Herndon JM, Eberlein TJ, Goedegebuure P, et al. Induction of Th17 cells in the tumor microenvironment improves survival in a murine model of pancreatic cancer. J Immunol. 2010;185(7):4063–4071. doi:10.4049/jimmunol.0902609.
- Kryczek I, Wei S, Szeliga W, Vatan L, Zou W. Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood. 2009;114:357–359. doi:10.1182/blood-2008-09-177360.
- Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009;31(5):787–798. doi:10.1016/j.immuni.2009.09.014.
- Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, Paulos CM, Palmer DC, Touloukian CE, Ptak K, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112(2):362–373. doi:10.1182/blood-2007-11-120998.
- Higashi T, Takagi R, Hashimoto K, Matsushita S. Curdlan induces dendritic cell-mediated Th17 polarisation via jagged1 activation in human dendritic cells. Allergy. 2010;65:339.
- Mori A, Oleszycka E, Sharp FA, Coleman M, Ozasa Y, Singh M, O’Hagan DT, Tajber L, Corrigan OI, McNeela EA, et al. The vaccine adjuvant alum inhibits IL-12 by promoting PI3 kinase signaling while chitosan does not inhibit IL-12 and enhances Th1 and Th17 responses. Eur J Immunol. 2012;42(10):2709–2719. doi:10.1002/eji.201242372.
- Werninghaus K, Babiak A, Gross O, Holscher C, Dietrich H, Agger EM, Mages J, Mocsai A, Schoenen H, Finger K. Adjuvanticity of a synthetic cord factor analogue for subunit Mycobacterium tuberculosis vaccination requires FcRgamma-Syk-Card9-dependent innate immune activation. J Exp Med. 2009;206:89–97. doi:10.1084/jem.20081445.
- Gasse P, Riteau N, Vacher R, Michel ML, Fautrel A, Di Padova F, Fick L, Charron S, Lagente V, Eberl G, et al. IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PLoS One. 2011;6. doi:10.1371/journal.pone.0023185.
- Preville X, Ladant D, Timmerman B, Leclerc C. Eradication of established tumors by vaccination with recombinant Bordetella pertussis adenylate cyclase carrying the human papillomavirus 16 E7 oncoprotein. Cancer Res. 2005;65:641–649.
- Lin KY, Guarnieri FG, Staveley-O’Carroll KF, Levitsky HI, August JT, Pardoll DM, Wu TC. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–26.
- Schoenen H, Bodendorfer B, Hitchens K, Manzanero S, Werninghaus K, Nimmerjahn F, Agger EM, Stenger S, Andersen P, Ruland J, et al. Cutting edge: mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J Immunol. 2010;184:2756–2760. doi:10.4049/jimmunol.0904013.
- Kim HS, Park KH, Lee HK, Kim JS, Kim YG, Lee JH, Kim KH, Yun J, Hwang BY, Hong JT, et al. Curdlan activates dendritic cells through dectin-1 and toll-like receptor 4 signaling. Int Immunopharmacol. 2016;39:71–78. doi:10.1016/j.intimp.2016.07.013.
- Schlecht G, Loucka J, Najar H, Sebo P, Leclerc C. Antigen targeting to CD11b allows efficient presentation of CD4+ and CD8+ T cell epitopes and in vivo Th1-polarized T cell priming. J Immunol. 2004;173:6089–6097. doi:10.4049/jimmunol.173.10.6089.
- Ma SB, Cheng Q, Cai YF, Gong HL, Wu Y, Yu X, Shi L, Wu D, Dong C, Liu H. IL-17A produced by gamma delta T cells promotes tumor growth in hepatocellular carcinoma. Cancer Res. 2014;74:1969–1982. doi:10.1158/0008-5472.CAN-13-2534.
- Fabre J, Giustiniani J, Garbar C, Antonicelli F, Merrouche Y, Bensussan A, Bagot M, Al-Dacak R. Targeting the tumor microenvironment: the protumor effects of IL-17 related to cancer type. Int J Mol Sci. 2016;17:E1433.
- Martin-Orozco N, Chung Y, Chang SH, Wang YH, Dong C. Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells. Eur J Immunol. 2009;39:216–224. doi:10.1002/eji.200838475.
- Hamai A, Pignon P, Raimbaud I, Duperrier-Amouriaux K, Senellart H, Hiret S, Douillard J-Y, Bennouna J, Ayyoub M, Valmori D. Human T(H)17 immune cells specific for the tumor antigen MAGE-A3 convert to IFN-gamma-secreting cells as they differentiate into effector T cells in vivo. Cancer Res. 2012;72:1059–1063. doi:10.1158/0008-5472.CAN-11-3432.
- Gagliani N, Vesely MCA, Iseppon A, Brockmann L, Xu H, Palm NW, de Zoete MR, Licona-Limón P, Paiva RS, Ching T, et al. TH17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. 2015;523:221–225. doi:10.1038/nature14452.
- Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64–70. doi:10.1189/jlb.0407247.
- Ng LG, Ostuni R, Hidalgo A. Heterogeneity of neutrophils. Nat Rev Immunol. 2019;19:255–265. doi:10.1038/s41577-019-0141-8.
- Schwarzenberger P, Huang WT, Ye P, Oliver P, Manuel M, Zhang ZL, Bagby G, Nelson S, Kolls JK. Requirement of endogenous stem cell factor and granulocyte-colony-stimulating factor for IL-17-mediated granulopoiesis. J Immunol. 2000;164(9):4783–4789. doi:10.4049/jimmunol.164.9.4783.
- Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, Wang Z, Wang C, Zhang Z, Xia W, et al. gamma delta T17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity. 2014;40:785–800. doi:10.1016/j.immuni.2014.03.013.
- Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5:3–8. doi:10.1158/2326-6066.CIR-16-0297.
- Zhou J, Nefedova Y, Lei AH, Gabrilovich D. Neutrophils and PMN-MDSC: their biological role and interaction with stromal cells. Semin Immunol. 2018;35:19–28. doi:10.1016/j.smim.2017.12.004.
- Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, Verstegen NJM, Ciampricotti M, Hawinkels LJAC, Jonkers J, et al. IL-17-producing gamma delta T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522:345–348. doi:10.1038/nature14282.
- Vono M, Lin A, Norrby-Teglund A, Koup RA, Liang F, Lore K. Neutrophils acquire the capacity for antigen presentation to memory CD4(+) T cells in vitro and ex vivo. Blood. 2017;129:1991–2001. doi:10.1182/blood-2016-10-744441.
- Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CPM, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079–1084. doi:10.1126/science.aad1329.
- Sandoval F, Terme M, Nizard M, Badoual C, Bureau MF, Freyburger L, Clement O, Marcheteau E, Gey A, Fraisse G, et al. Mucosal imprinting of vaccine-induced CD8(+) T cells is crucial to inhibit the growth of mucosal tumors. Sci Transl Med. 2013;5:172ra20.
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi:10.1016/j.cell.2009.09.033.
- Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, Laufer T, Ignatowicz L, Ivanov I. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity. 2014;40(4):594–607. doi:10.1016/j.immuni.2014.03.005.
- Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gamma delta T cells rather than CD4 T cells during mycobacterium tuberculosis infection. J Immunol. 2006;177:4662–4669. doi:10.4049/jimmunol.177.7.4662.
- Flannigan KL, Ngo VL, Geem D, Harusato A, Hirota SA, Parkos CA, Lukacs NW, Nusrat A, Gaboriau-Routhiau V, Cerf-Bensussan N, et al. IL-17A-mediated neutrophil recruitment limits expansion of segmented filamentous bacteria. Mucosal Immunol. 2017;10:673–684. doi:10.1038/mi.2016.80.
- Reppert S, Boross I, Koslowski M, Tureci O, Koch S, Lehr HA, Finotto S. A role for T-bet-mediated tumour immune surveillance in anti-IL-17A treatment of lung cancer. Nat Commun. 2011;2:600.
- Ma YF, Chen C, Li DQ, Liu M, Lv ZW, Ji YH, Xu J. Targeting of interleukin (IL)-17A inhibits PDL1 expression in tumor cells and induces anticancer immunity in an estrogen receptor-negative murine model of breast cancer. Oncotarget. 2017;8:7614–7624. doi:10.18632/oncotarget.13819.