
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Antigen-encoding, lipoplex-formulated RNA (RNA-LPX) enables systemic delivery to lymphoid compartments and selective expression in resident antigen-presenting cells. We report here that the rejection of CT26 tumors, mediated by local radiotherapy (LRT), is further augmented in a CD8+ T cell-dependent manner by an RNA-LPX vaccine that encodes CD4+ T cell-recognized neoantigens (CD4 neoantigen vaccine). Whereas CD8+ T cells induced by LRT alone were primarily directed against the immunodominant gp70 antigen, mice treated with LRT plus the CD4 neoantigen vaccine rejected gp70-negative tumors and were protected from rechallenge with these tumors, indicating a potent poly-antigenic CD8+ T cell response and T cell memory. In the spleens of CD4 neoantigen-vaccinated mice, we found a high number of activated, poly-functional, Th1-like CD4+ T cells against ME1, the immunodominant CD4 neoantigen within the poly-neoantigen vaccine. LRT itself strongly increased CD8+ T cell numbers and clonal expansion. However, tumor infiltrates of mice treated with CD4 neoantigen vaccine/LRT, as compared to LRT alone, displayed a higher fraction of activated gp70-specific CD8+ T cells, lower PD-1/LAG-3 expression and contained ME1-specific IFNγ+ CD4+ T cells capable of providing cognate help. CD4 neoantigen vaccine/LRT treatment followed by anti-CTLA-4 antibody therapy further enhanced the efficacy with complete remission of gp70-negative CT26 tumors and survival of all mice. Our data highlight the power of combining synergistic modes of action and warrants further exploration of the presented treatment schema.
Introduction
Half of the patients with cancer irrespective of tumor type undergo local radiotherapy for curative or palliative purposes.Citation1 In addition to its anti-proliferative and cytotoxic activity on cancer cells, LRT mediates a multitude of effects. These include the induction of immunogenic cell death, release of danger-associated molecular patterns, activation of antigen-presenting cellsCitation2-Citation4 and may promote the priming and effector phase of tumor-reactive T cells. As a consequence, LRT is considered to convert the tumor to an in situ vaccine,Citation5 which is the rationale for combining LRT with immune modulators such as antibodies against CTLA-4,Citation6 PD-1/PD-L1,Citation7 CD40,Citation8 or CD137Citation9 and also with CD8+ T cell-inducing cancer vaccines.Citation10–Citation12
We have previously reported a comprehensive cancer mutanome analysis of mouse tumors showing that a considerable fraction of non-synonymous cancer mutations are immunogenic, that the majority of the immunogenic mutanome is recognized by CD4+ T cells and that vaccination with such CD4+ T cell-reactive immunogenic mutations confers strong antitumor activity.Citation13 In this regard, we were specifically interested to study high dose LRT in conjunction with a vaccine inducing tumor neoantigen-specific CD4+ T cells.
To this aim, we resorted to a cancer vaccine model based on the CT26 colon carcinoma in BALB/c mice. In this model, a pentatope vaccine (CT26 PME1), engineered from five highly expressed CT26-specific mutations (`monotopes`) with strong predicted major histocompatibility complex (MHC) class II binding capacity, mediates complete rejection in an aggressively growing hematogenic dissemination simulating lung metastasis model of CT26,Citation13 whereas it is ineffective against subcutaneously (s.c.) established CT26 tumors.
The tumor rejection depends on cytotoxic CD8+ T cells including specificities against gp70-AH1,Citation14 the immunodominant gp70-epitope in CT26. The pentatope vaccine induces a poly-epitopic CD4+ T cell response, with CT26 ME1 (Aldh18a1P154S), being the most immunogenic CT26 PME1-encoded CD4 neoantigen.Citation13 The vaccine format used in this model is a single-stranded antigen-encoding RNA encapsulated in liposomes (RNA-LPX).Citation15 The RNA has been engineered for optimized intracellular stability and translational efficiencyCitation16-Citation18 and for augmented presentation not only on MHC class I but also MHC class II.Citation19 Intravenously (i.v.) administered RNA-LPX target to lymphoid compartments and are taken up and expressed exclusively by resident antigen-presenting cells.Citation15 As a natural TLR7/8 ligand, RNA mediates a strong type I interferon (IFN) dominated innate response, concurrent to delivering the encoded antigen.Citation15,Citation20,Citation21 The described mode of action in mice is supported by preliminary observations in ongoing clinical trials with RNA-LPX in patients with solid cancers.Citation21–Citation24
The purpose of the study presented here was to utilize the described mouse model to investigate whether a CD4 neoantigen vaccine can synergize with LRT and to characterize the involved mechanisms.
Our data indicate that CD4 neoantigen vaccination maximizes radiation-induced adaptive T cell responses by boosting in situ CD8+ T cell immunity.
Materials and methods
Mice
BALB/c wild-type mice were purchased from Janvier and age-matched (8–12 weeks) female animals used throughout all experiments. Procedures and experimental group sizes were approved by the regulatory authorities for animal welfare. All mice were kept in accordance with federal and state policies on animal research at BioNTech SE.
Tumor cell lines
The murine BALB/c colorectal cancer cell line CT26 was purchased from ATCC (CRL-2638, lot no. 58494154). CT26 cells present the immunodominant gp70 antigen, which is a viral envelope protein endogenously expressed in BALB/c mice, but silent in most normal mouse tissues.Citation25 The gp70-epitope AH1 (SPSYVYHOF) is known to elicit strong CD8+ T cell responses in BALB/c mice.Citation14 CT26-gp70KO cells were generated via CRISPR/Cas9 mediated introduction of indels into the gp70 locusCitation26 and thus not recognized by gp70-AH1-specific splenocytes. The murine BALB/c 4T1-luc2-tdTomato (4T1-luc) breast cancer cell line was purchased from Caliper Life Sciences (125669, lot no. 101648). Master and working cell banks were generated immediately upon receipt/generation. Cells from fifth to ninth passage were used for in vivo tumor experiments. Cells were tested for mycoplasma contamination every 3 months.
Mutation selection
For mutation detection, RNA and whole-exome sequencing of CT26 and 4T1-luc tumor cells and BALB/c tail tissue samples was performed by TRON gGmbH (Mainz, Germany) as earlier described.Citation13 Sequencing FASTQ files for CT26, 4T1-luc, and BALB/c are available from the European Nucleotide Archive (ENA) as PRJEB5321, PRJEB5320, PRJEB5791, and PRJEB5797. The computational pipeline for identification of immunogenic CT26 neoantigens was reported previously.Citation13 For mutation detection, Strelka (version 2.0.14)Citation27 and VarScan2 (version 2.3.5)Citation28 were used, and for MHC-binding prediction, the IEDB consensus method version 2.12. The 4T1 pentatope, Pctrl. was selected as a control pentatope for experiments with pentatope CT26 PME1Citation13 and features immunogenic cancer mutations of the BALB/c 4T1 mouse tumor model, which are not present in BALB/c CT26 tumor models. As previously reported, of the CT26 PME1 encoded mutations, CT26 ME1 and ME2 (Supplementary Figure 2(b)) triggered antigenic CD4+ T cell responsesCitation13 and of the Pctrl. encoded mutations 4T1 M53, M56, and M57 triggered antigenic CD4+ T cell responses (Supplementary Figure 2(a)).
RNA constructs and in vitro transcription
Plasmid templates for in vitro transcription of antigen-encoding RNAs were based on the pST1-A120 and pST1-MITD vector which feature 5` and 3`UTRs and poly(A) tails pharmacologically optimized for stability and protein translation.Citation17 pST1-MITD features a signal sequence for routing to the endoplasmic reticulum and MHC class I transmembrane and cytoplasmic domains for improved presentation of MHC class I and II epitopes.Citation19 Mutated amino acids were encoded as 27-mers containing the non-synonymous single nucleotide variants (nsSNV) at position 14 and embedded into pST1-MITD vectors as pentatopes, fused to each other by 10 amino acid long glycine-serine linkers or as monotopes. The following antigen-encoding vectors were used: CT26 PME1 (pentatope, encoding the CT26 neoantigens Aldh18a1P154S (ME1), Ubqln1A62V (ME2), Ppp6r1D309N (ME3), Trip12V1328M (ME4), Pcdhgc3E139K (ME5)),Citation13 CT26 ME1 (monotope, encoding the CT26 neoantigen Aldh18a1P154S (ME1)); Pctrl. (pentatope, encoding the 4T1-luc neoantigens GnpatS110R (M53), Isoc1V205L (M54), H2-Q6E179D (M55), Chsy1G459A (M56), Pi4kbT17A (M57)). Antigen-encoding vectors were in vitro transcribed and RNA capped with β-S-anti-reverse cap analog (ARCA) as previously described.Citation17,Citation29 As control RNA, the same vector backbone without an antigen-encoding open reading was used.
RNA-LPX preparation
RNA-LPX were generated by complexing RNA with liposomes consisting of the cationic lipid DOTMA and the helper lipid DOPE as previously described.Citation15 In brief, RNA-LPX were prepared using RNA stored in HEPES-buffered solution (1 mg mL−1) diluted with H2O and 1.5 M NaCl and adding the liposome dispersion to reach a charge ratio of (+):(-) 1.3:2 and a final NaCl concentration of 150 mM. RNA-LPX preparations had a particle size of ~340 nm and a polydispersity index of ~0.4.
Synthetic peptides
Peptides containing non-synonymous single nucleotide variants were synthetized as 27-mer peptides, containing the mutated amino acid at center position (underlined),Citation13 CT26 ME1 (Aldh18a1P154S, peptide sequence LHSGQNHLKEMAISVLEARAC AAAGQS), CT26 ME2 (Ubqln1A62V, DTLSAMSNPRAMQVLLQIQQGLQTLAT), 4T1 M53 (GnpatS110R, VLREEASEILEEMRHKLRIGAIRFFAF), 4T1 M54 (Isoc1V205L, EAALAEIPGVRSVLLFGVETHVCIQQT), 4T1 M55 (H2-Q6E179D, PADITLTWQLNGE DLTQDMELVETRPA), 4T1 M56 (Chsy1G459A, YILDLLLLYKKHKAKKMTVPVRRHAYL) and 4T1 M57 (Pi4kbT17A, GKRLATLPTKEQKAQRLIS ELSLLNHK). The gp70-AH1 (gp70423-431) peptide with a single amino acid substitution at position five (V427A) was synthetized as 9-mer (peptide sequence SPSYAYHQF).Citation14 JPT Peptide Technologies GmbH synthesized all peptides with a purity above 70%.
Tumor models and treatment
For therapeutic tumor experiments, BALB/c mice were injected with 5 × 105 CT26 or 3 × 105 CT26-gp70KO tumor cells subcutaneously to the right flank. Tumor growth was measured unblinded with a caliper every 3 to 4 days and tumor volumes calculated by (a2 × b)/2 (a, width; b, length). Tumor growth is displayed on a nonlinear, logarithmic scale (log2) allowing the better visualization of tumor growth kinetics, especially at smaller tumor volumes. Mice were immunized with 40 µg RNA-LPX intravenously every 5 to 7 days. Local tumor irradiation was performed using the orthovoltage X-ray source X-Rad320 from Precision X-Ray Inc. at a dose rate of 0.47 Gy/min applying either 12 Gy or an equivalent biologically effective dose (BED) of 3 × 6 Gy. Biological effective doses were calculated using the formula with n being the number of fractions, d being the dose per fraction and being the tumor intrinsic radio-sensitivity.Citation30 Tumor irradiation was performed under ketamine/xylazine (12 Gy) or isoflurane (3 x 6 Gy) narcosis. Mice were completely shielded by a custom-made lead shield that spares a hole (1.5 cm diameter) allowing exposition only of the tumor tissues. Prior radiotherapy mice were randomized according to their tumor volume and were eligible when tumors exhibited volumes between 13,5 mm3 – 200 mm3. Anti-CD8 (YTS169.4, BioXCell) depleting antibody was injected intraperitoneal at 200 µg every 3 to 4 days. Anti-CTLA-4 (9H10, BioXCell) and anti-PD-1 (RMPI-14, BioXCell) antibodies were injected intraperitoneal thrice, at 200 µg (first) and 100 µg (second and third), every 5 days. For tumor rechallenge experiments, mice were inoculated with 3 × 105 CT26-gp70KO tumor cells to the opposite (left) flank. Animals were euthanized when exhibiting signs of impaired health or when the tumor volume exceeded 1500 mm3.
Tissue preparation
For the generation of single-cell suspensions, tumors were cut and digested using the mouse tumor dissociation kit and gentleMACSTM dissociator (Miltenyi Biotec) and were forced through a 70 µm cell strainer (BD Falcon), using a plunger end of a syringe while rinsing with PBS. Cells were centrifuged at 460 × g for 6 min and resuspended in fresh PBS. Erythrocytes were lysed with a hypotonic electrolyte solution for 5 min. Similarly, spleens were forced through a 70 µm cell strainer while rinsing with PBS and erythrocytes lysed with hypotonic electrolyte solution. For the isolation of tumor CD45+ or CD8+ cells, target cells were magnetically enriched using the mouse CD8 (TIL) or CD45 (TIL) MicroBeads and for spleen CD4+ or CD8+ T cells using the CD4 (L3T4) or CD8a (Ly-2) MicroBeads (Miltenyi Biotech) according to manufacturer’s instructions. Peripheral blood was collected from the orbital sinus for flow cytometry staining (50 µl), TCRαβ sequencing (100 µl) and blood IFNγ ELISpot (120 µl). For blood IFNγ ELISpot, blood lymphocytes were enriched via ficoll (Ficoll®-Paque PREMIUM 1.084) gradient according to the manufacturer’s instructions, pooling the blood of 3–4 mice.
Flow cytometry
Flow cytometry staining was conducted on full blood, tumor, and spleen single-cell suspension as well as on CD45+ T cell-enriched tumor samples and CD4+ T cell-enriched spleen samples. Monoclonal antibodies for extracellular staining included CD4, CD8a, CD40L, CD44, CD45, CD69, TIGIT (BD Pharmingen), LFA-1, PD-1 (BioLegend), CD25, CD62L, ICOS, Lag-3, Tim3 (eBioscience). For intracellular staining, antibodies against IFNγ (Invitrogen) and CD40L, T-bet, TNFα (BD Pharmingen) were used. Live cells were stained with viability dyes (eBioscience) according to the manufacturer’s instructions. Gp70-AH1-specific CD8+ T cells were stained with gp70423-431 H2-Ld-restricted tetramers (MBL) for 10 min at 4°C in the dark. ME1-specific CD4+ T cells were stained after CD4+ T cell MACS enrichment with 12 µg/mL custom-made CT26 ME1 I-A(d)-restricted tetramers (peptide sequence NHLKEMAISVLEARA, from NIH Tetramer Core Facility) for 45 min at room temperature in the dark. Full blood was stained with gp70423-431 H2-Ld-restricted tetramers prior to erythrocyte lysis using BD FACS lysing solution (BD Pharmingen). Extracellular targets were stained for 30 min at 4°C in the dark. PBS containing 5% FCS and 5 mM EDTA were used as washing and staining buffer. For the intracellular staining of IFNγ, TNFα, and CD40L, samples were fixed and permeabilized with Cytofix/Cytoperm (BD Pharmingen), whereas for T-bet staining, samples were fixed and permeabilized using Foxp3 Fixation Kit (eBioscience) according to manufacturer’s instructions. Intracellular cytokine staining was performed as earlier described,Citation31 stimulating 5 × 106 CD45 MACS TILs with 5 × 104 2 µg/mL gp70-AH1 or 5 × 104 5 µg/mL ME1 peptide-loadedCitation32 BALB/c bone marrow-derived cells (BMDC) for 5 h at 37°C in the presence of 10 µg/mL Brefeldin A (Sigma). Immune cell populations were defined by pre-gating on viable cells and singlets and determined as follows: CD8+ T cells (CD45+ CD8+), gp70-specific CD8+ T cells (CD45+ CD8+ gp70-AH1 tetramer+), CD4+ T cells (CD45+ CD4+), ME1-specific CD4+ T cells (CD45+ CD4+ ME1 tetramer+ or CD45+ CD4+ CD40L+ after ME1-peptide restimulationCitation32), tumor cells (CD45− CD44+). Flow cytometric data were acquired with LSR Fortessa flow cytometer (BD Biosciences) and analyzed with FlowJo 10.4 software (Tree Star).
IFNγ ELISpot
Enzyme-linked ImmunoSpot assay (ELISpot) was performed as previously described to detect T cell IFNγ release upon antigen encounter.Citation33 In brief, 1 × 105 MACS enriched CD8+ T cells were restimulated with 5 × 104 2 µg/mL peptide-loaded BALB/c BMDC or 5 × 104 tumor cells, respectively, and 5 × 105 MACS enriched CD4+ T cells with 5 × 104 5 µg/mL peptide-loaded BALB/c BMDC overnight at 37°C. In supplementary Figure 2(a), 5 × 105 CD8+ T cell-depleted splenocytes were used as effector cells. IFNγ spots of ficoll purified blood lymphocytes were normalized to CD8+ T cell counts determined retrospectively via flow cytometry using CountBrightTM Absolute Counting Beads (ThermoFisher Scientific). In some instances, tumor cells were irradiated with 20 Gy (X-Rad320, Prevision X-Ray Inc.) 24 h prior to co-incubation with target cells as maximum MHC class I upregulation was observed at this dose (Supplementary Figure 3 a). All samples were tested in duplicates or triplicates.
TCR sequencing and analysis
Bulk αβTCR sequencing was performed on cryo-conserved blood and intratumoral CD8+ T cells after magnetic separation (MACS®, Miltenyi Biotec). Total RNA was extracted from blood followed by clean up on QIAcube using RNeasy mini spin columns. RNA was reverse-transcribed to cDNA using SMARTer Mouse TCR a/b Profiling kit. Bulk αβTCR sequencing was performed on MiSeq using MiSeq Reagent Kit v3. Raw sequencing data were aligned and TCR sequences obtained using MiXCR.Citation34 The data were further processed using VDJtoolsCitation35 including frequency-based error correction and filtering of nonfunctional sequences. Metrics were calculated for the intratumoral CD8+ T cell TCRβ counts with richness as the number of unique clones found within the sample and clonality being the mean clonotype frequency. The fraction of blood-tumor shared TCRβ clones was determined by: (no. of blood-tumor shared clones)/(no. of all tumor clones) x 100.
Multiplex immunoassay
Cell culture supernatants from peptide-restimulated CD4+ T cells were analyzed using the Th1/Th2/Th9/Th17/Th22/Treg cytokine 17-Plex mouse ProcartaPlexTM immunoassay (ThermoFisher Scientific). Mice were vaccinated weekly with RNA-LPX, spleens excised 5 days after the third immunization and CD4+ T cells magnetically enriched (MACS®, Miltenyi Biotec). 5 × 105 CD4+ T cells were co-incubated with 1 × 105 10 µg/mL ME1 peptide-loaded BMDC for 48 h at 37°C. Data was acquired using the Bioplex200 (Bio-Rad) and analyzed with ProcartaPlexAnalyst software. All samples were tested in duplicates.
Statistical analysis
Single treatment and control group means were compared by unpaired, two-tailed Student’s t-test. If more than two experimental groups were compared, one-way analysis of variance (ANOVA) was performed and when determined significant (p < 0.05), Tukey’s multiple comparison test run. Survival benefit was determined using log-rank test (Mantel-Cox). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. If not mentioned otherwise, results are depicted as mean±SEM. All statistical analyses were performed with GraphPad PRISM 8.
Results
A CD4 neoantigen vaccine improves LRT-mediated survival of mice with CT26 tumors in a CD8+ T cell-dependent manner
To extend the model system described above for testing of the CT26 PME1 RNA-LPX-based CD4 neoantigen vaccine/LRT combination, we first assessed two LRT protocols. We treated s.c. CT26 tumors of BALB/c mice with LRT, applying either three doses of 6 Gy at consecutive days or one equivalent biologically effective dose of 12 Gy. Both LRT regimens resulted in tumor growth inhibition and improved survival of mice with only occasional observations of tumor regression ). Both protocols were equally effective in inducing a strong adaptive CD8+ T cell response against gp70-AH1, the immunodominant antigen of the CT26 tumor model ). We chose single dose over the fractionated irradiation for practical reasons for further experiments.
We adapted the CT26 vaccine model described above (subcutaneous CT26 tumors and a sub-therapeutic prime/boost schema) to obtain a setting in which the vaccine alone would not be effective. We administered three cycles of the CD4 neoantigen vaccine and LRT in between the second and third RNA-LPX vaccination ). While LRT alone had a marginal antitumor effect, its combination with the CD4 neoantigen vaccine increased the rate of complete responses by 6-fold ) and significantly improved survival ). Expansion of gp70-specific CD8+ T cells in blood was achieved with different kinetics by each of the individual components and it was significantly more efficient and sustained upon combining the CD4 neoantigen vaccine to LRT ). Again, the same findings were made when 12 Gy LRT was fractionated to an equivalent biological dose of 3 × 6 Gy (Supplementary Figure 1(a, b)).
Depletion of CD8+ T cells by systemic administration of an anti-CD8 antibody (aCD8) after the CD4 neoantigen vaccine/LRT treatment eradicated circulating gp70-AH1 specific CD8+ T cells ) and significantly diminished tumor rejection and survival benefits in mice treated with the combination ).
Activated poly-functional Th1-like CD4+ T cells against the immunodominant vaccine-encoded CD4 neoantigen are induced in the spleen of CD4 neoantigen vaccine/LRT treated mice
First, we wanted to characterize the neoantigenic CD4+ T cell response as the most direct effect of the CD4 neoantigen vaccine to understand its contribution to the CD4 neoantigen vaccine/LRT-mediated synergy.
To investigate if CD4 neoantigen vaccine/LRT-mediated therapeutic effects rely on multiple neoantigen-specific CD4+ T cell responses or if a single, immunodominant, CD4+ T cell neoantigen could confer similar antitumor effects, the pentatopic CT26 PME1 version of the CD4 neoantigen vaccine was compared to CT26 ME1, the immunodominant CT26 PME1-encoded antigen (Supplementary Figure 2(b)). Similar therapeutic effects were reached when LRT was combined with either CT26 PME1 or CT26 ME1 RNA-LPX ). In addition, the need of CD4+ T cell tumor-specificity was assessed, combining LRT with a tumor irrelevant CD4 neoantigen vaccine (Pctrl. RNA-LPX), encoding CD4+ T cell-reactive neoantigens identified in the 4T1 mouse breast cancer model and thus termed irrelevant in the context of CT26. Pctrl. RNA-LPX induced poly-epitopic IFNγ+ CD4+ T cells (Supplementary Figure 2(a)), but did not augment the antitumor effect of LRT in CT26 tumors ), indicating that CD4+ T cells contribute to the CD4 neoantigen vaccine/LRT-mediated synergy by providing cognate help.
We vaccinated tumor-naïve, non-irradiated mice with the CD4 neoantigen vaccine and characterized vaccine-induced CD4+ T cells in the spleen ), the major compartment with expression of RNA-LPX-encoded antigens.Citation15 We stained CD4+ T cells with tetramers against the CT26 ME1 monotope, which we had confirmed to contribute dominantly to tumor rejection, survival benefit, and gp70-specific CD8+ T cell expansion after combined LRT ). ME1-specific CD4+ T cells of PME1 RNA-LPX vaccinated mice displayed an activated effector phenotype with upregulation of ICOS, CD69, and PD-1 and downregulation of CD62L ), which was not the case for splenocytes of control RNA-LPX vaccinated mice or of the tetramer-negative fraction of vaccine-treated mice. ME1-peptide restimulated bulk splenocytes showed expression of the transcription factor T-bet ), together with the dominant secretion of cytokines such as IFNγ, IL-2, IL-18, IL-12p70, TNFα, IL-1β ), indicating enrichment of epitope-specific CD4+ T cells with a Th1-like phenotype.Citation36
In addition, CT26 tumor-bearing mice treated with the CD4 neoantigen vaccine/LRT had a strong increase of splenic CD40L-positive CD4+ T cells recognizing the immunodominant ME1 antigen on peptide-pulsed target cells ).
Adding the CD4 neoantigen vaccine to LRT results in a potent polyantigenic CD8+ T cell response and T cell memory
Having shown that synergistic antitumor effect of the CD4 neoantigen vaccine/LRT combination depends on the expansion of CD8+ T cells ), we investigated the CD8+ T cell response in more detail.
Testing splenocytes of CT26 tumor-bearing mice in IFNγ ELISpot confirmed that CD8+ T cells induced by either LRT alone or by its combination with vaccine were largely directed against gp70-AH1. Recognition of gp70-knock-out CT26 tumor cells (CT26-gp70KO, including irradiated ones to reflect the tumor setting after LRT, with MHC upregulationCitation37) was more pronounced for CD8+ T cells of combination-treated mice, with a 3-fold higher mean spot number as compared to LRT alone ).
To analyze the functional importance of non-gp70-directed CD8+ T cell responses, the therapeutic efficacy of the CD4 neoantigen vaccine/LRT combination was assessed in CT26-gp70KO tumor models ). The CD4 neoantigen vaccine/LRT combination mediated complete tumor rejection in nearly half of CT26-gp70KO tumor-bearing mice and resulted in significantly superior survival, whereas either of the single modalities was ineffective ). This effect was associated with expansion of CD8+ T cells in the spleen that recognized CT26-gp70KO target cells in IFNγ ELISpot ).
In line with this observation, mice that previously rejected advanced CT26 tumors under CD4 neoantigen vaccine/LRT treatment remained tumor-free after rechallenge with CT26-gp70KO cells, indicating protective immunity against this otherwise lethal tumor challenge ).
In conclusion, these data strongly suggest that adding a tumor-specific CD4 neoantigen vaccine to LRT induces a gp70 AH1-independent, polyantigenic and memory-type cytotoxic CD8+ T cell response.
Tumor immune infiltrates of CD4 neoantigen vaccine/LRT-treated mice contain activated, tumor antigen-specific CD8+ T cells and vaccine-induced CD4+ T cells capable of providing cognate help
When characterizing tumor-infiltrating lymphocytes, we found that LRT alone significantly increased the fraction of total leukocytes and CD8+ T cells and reduced the fraction of tumor cells within the CT26 tumors and the vaccine did not further add to these effects ). In contrast, LRT alone did not affect the frequency of intratumoral gp70-AH1 specific CD8+ T cells within CD8+ T cells, whereas this cell fraction was significantly increased when adding the CD4 neoantigen vaccine ).
Intratumoral gp70-AH1-specific CD8+ T cells from CD4 neoantigen vaccine/LRT treated mice displayed significantly lower levels of immunosuppressive markers PD-1 and Lag-3 and higher levels of the transmigration marker LFA-1 ). Further, tumor-infiltrating CD8+ T cells from combination-treated mice contained a significantly higher number of poly-functional IFNγ/TNFα-secreting gp70-AH1-specific effector CD8+ T cells ).
Next, we aimed to characterize the intra-tumoral CD8+ T cell repertoire and performed TCRβ sequencing of CD8+ tumor-infiltrating lymphocytes (TILs) from CT26 tumor-bearing mice 9 days after LRT ). LRT reduced the number of unique TCRβ clones (richness) and increased TCRβ clonality, indicating skewing of the polyclonal TCRβ repertoire toward defined clonotypes. The fraction of blood-tumor-shared clones increased after LRT, implying that LRT induces new CD8+ T cell clones, which are able to circulate and reach the tumor site ).
With regard to tumor-infiltrating CD4+ T cells, whereas the overall fraction was not affected by any of the treatments ), CD4+ T cells capable of recognizing CT26 ME1-peptide-pulsed target cells were found only within tumors of combination-treated mice. ME1-peptide restimulated CD4+ T cells from CD4 neoantigen vaccine/LRT treated mice were strongly CD40L- and IFNγ-double positive ), indicating the presence of cognate help at the tumor site.
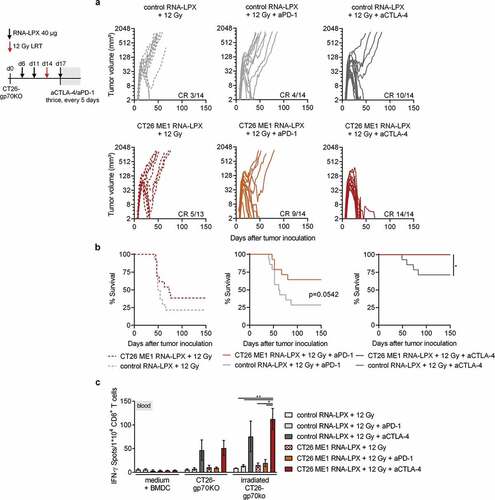
CD4 neoantigen vaccine/LRT treatment followed by anti-CTLA4 antibody therapy further enhances the efficacy with complete remission of gp70-negative CT26 tumors and survival of all mice
As the benefit of combining checkpoint inhibitors (CPIs) with high dose LRT has previously been shown,Citation38 we wondered whether CPIs could further augment the synergistic antitumor effect of the CD4 neoantigen vaccine/LRT combination.
We chose the harder to treat CT26-gp70KO tumor model in combination with the CT26 ME1 monotope version of the CT26 PME1 CD4 neoantigen vaccine. LRT was again administered between the second and third vaccine, dosing at a slightly larger tumor volume (~60 mm3) than in previous experiments, and either anti-CTLA-4 or anti-PD-1 treatment started concomitantly with the last cycle of the CD4 neoantigen vaccine (). Adding CPIs to the CD4 neoantigen vaccine/LRT combination had a profound effect on tumor rejection and survival. In particular, the CD4 neoantigen vaccine/LRT/anti-CTLA-4 combination resulted in the rejection of all tumors and survival of all mice ). The fraction of circulating tumor-reactive T cells detected by IFNγ ELISpot in the CD4 neoantigen vaccine/LRT/anti-CTLA-4 triple-treated group was significantly higher as compared to LRT and LRT/anti-PD-1 with or without the CD4 neoantigen vaccine ).
Discussion
Immune-stimulatory properties of high dose LRT are increasingly recognized; hence, new combination regiments are designed that aim to maximize LRT-induced in situ immune responses.Citation39 As a majority of the immunogenic mutanome is recognized by CD4+ T cells,Citation13 we set out to identify whether RNA-LPX vaccination against cognate CD4+ T cell neoantigens can synergize with LRT to enhance antitumoral effects in a murine cancer model.
Innate immune activation, type I IFN signaling and recruitment of cross-presenting DCs to the local tumor environment are decisive effects of high dose LRT to trigger tumor antigen-specific T cell responses.Citation4,Citation6,Citation40 These in situ vaccination effects have been observed in sporadic clinical case reports following LRT.Citation41 In mice, high dose LRT lead to the priming of SIY-specific CD8+ T cells,Citation42 OVA-specific CD4+, CD8,+ and TRP-2-specific CD8+ T cellsCitation43 in B16 melanomas engineered to overexpress these antigens.Citation42 We observed a similar effect in s.c. CT26 tumor-bearing mice, where 12 Gy LRT lead to the priming of gp70-AH1-specific CD8+ T cells seven to 10 days after LRT. Single, 12 Gy, LRT induced a more clonal T cell response in CT26 tumors and increased the number of blood-tumor-shared T cell clones, which was also observed for daily 2 Gy fractionated LRT in conjunction with anti-PD-L1 CPI in s.c. CT26 tumor models.Citation44 However, LRT alone was ineffective in rejecting s.c. CT26 tumors, which relapsed after a short phase of growth inhibition.
Different therapeutic strategies aim to maximize LRT in situ vaccination, modulating the radiation doseCitation45 as well as assessing the combination with different CPIsCitation6,Citation7,Citation38 or cancer vaccinesCitation12,Citation46,Citation47 to increase adaptive immune responses. While LRT dose-fractionation did not augment in situ vaccination effects in CT26 tumor models, vaccination with a cognate CD4+ T cell neoantigen did.
Therapeutic effects of LRT and cancer vaccines have been identified in some preclinical studies, mostly vaccinating against foreign, overexpressed CD8+ T cell antigens.Citation10,Citation12,Citation47 LRT-primed GUC2Y-specific CD8+ T cell responses were amplified by an adenoviral GUC2Y vaccine, rendering an otherwise ineffective vaccine therapeutically valuable.Citation12 In another study, the survival of E.G.7-OVA tumor-bearing mice was enhanced by fractionated LRT and OVA-specific RNA vaccines, triggering a CD8+ T cell response.Citation47 Such combination studies primarily leveraged LRT-mediated growth inhibition and inflammation to potentiate vaccine-induced CD8+ T cells responses. Differing from the above-mentioned approaches, we combined LRT with a RNA-LPX vaccine encoding tumor-specific CD4 neoantigens, aiming to compensate for the lack of potent cognate help during in situ vaccination.
Tumor-specific CD8+ T cells recognize and kill tumor cells directly, whereas CD4+ T cells are able to modulate tumor immunity in many ways.Citation48 They possess the ability to secrete inflammatory cytokines, directly kill MHC-II positive tumorsCitation49 or modulate antitumor T and B cell responses in magnitude and quality.Citation50,Citation51 The CD4 neoantigen vaccine itself was not able to reject MHC class II-negative s.c. CT26 tumors, but it strongly augmented the antitumoral efficacy of LRT in a CD8+ T cell-dependent manner. We identified Th1-like CD4+ T cells recognizing the cognate CT26 neoantigen ME1 (Aldh18a1P154S),Citation13 a member of the aldehyde dehydrogenase family,Citation52 to be solely responsible for therapeutic benefits observed together with LRT as vaccination with this neoantigen conferred same anti-tumoral effects as vaccination with five cognate CT26 neoantigens.
The CD4 neoantigen vaccine/LRT combination increased the fraction of antigen-specific CD8+ T cells within tumor-infiltrating lymphocytes, which was not achieved by LRT alone. Also, tumor-infiltrating antigen-specific CD8+ T cells had a less suppressed and more cytotoxic phenotype. Similar effects were reported in vaccination settings, physically linking helper CD4+ and CD8+ T cell epitopes for concomitant presentation of MHC-I and II antigens on cognate DC.Citation51 Also, on the axis of CD4 help, but with a slightly different approach, the combination of total body irradiation and agonistic anti-CD40 antibody was reported, which mediated superior rejection of mouse lymphoma models in a CD8+ T cell-dependent manner.Citation8 Detailed mechanisms of where and when CD4 neoantigen vaccine-induced T cells boost LRT-induced CD8+ T cell responses – i.e., via DC-licensing or via the secretion of additional cytokines in the TME – remain to be elucidated.
Whereas the CD4 neoantigen vaccine/LRT combination mainly induced CD8+ T cells reactive against the immunodominant antigen gp70-AH1, gp70-independent CD8 T cell responses were observed in CT26-gp70KO tumor models that were antitumorally active and protected from tumor rechallenge. These CT26-gp70KO antigens could arise from mutation, overexpression, or be cancer-germline antigens.Citation13,Citation26,Citation53
In preclinical and clinical research, LRT is most often combined with CPIs such as anti-PD-1/PD-L1 or anti-CTLA-4.Citation38 Antibodies blocking CTLA-4 have been reported to increase LRT-mediated priming, tipping the CD8+ T cell/Treg ratio,Citation5,Citation38 while combined anti-PD-1/PD-L1 treatment may release the “breaks” of T cell inhibition, enhancing the killing efficacy of primed CD8+ T cells.Citation38,Citation54,Citation55 When classical schedules of LRT/CPI were combined with the CD4 neoantigen vaccine, antitumoral responses were strengthened. This indicates that CPI and CD4 neoantigen vaccines potentially activate non-redundant mechanisms when combined with LRT.
To our knowledge, there are no previous reports describing the synergistic combination of LRT and CD4 neoantigen vaccines. Two studies reported the combination of LRT and adoptively transferred tumor-specific Th1-type CD4+ T cells against OVA323-339 in EG7Citation56 and TRP-1 in B16Citation57 tumor-bearing mice, but therapeutic effects of the CD4+ T cell component were rather modest.
Due to advances in next-generation sequencing and data science, comprehensive mapping and prediction of MHC-binding neoantigens is feasible.Citation58 The vast majority of neoantigens were recognized surprisingly by CD4+ rather than by CD8+ T cells.Citation13 Neoantigen-specific CD8+ T cells are able to kill tumor cells directly, putting selective pressure on the tumor and thus potentially deleting tumor clones expressing strong CD8 neoantigens during cancer evolution.Citation59 Neoantigen-specific CD4+ T cells on the other hand are not able to bind tumor cells, as they are mostly MHC class II negative; thus, CD4+ T cell-reactive neoantigens may be less prone to immunoediting and more ubiquitously represented within the tumor. We herein present a novel vaccination strategy, utilizing LRT to prime tumor-specific T cell responses while our CD4 neoantigen vaccine further expands the full repertoire of tumor-relevant T cell clones by cognate T cell help. This vaccination strategy is of special interest, i.e. when MHC class I restricted antigens are absent, sub-clonal, or non-identifiable.
Our data highlight the importance of neoantigen-specific CD4+ T cells to establish potent in situ T cell immunity after LRT and warrants further efforts to validate the combinatorial regimen in clinical trials.
Figure 1. A CD4 neoantigen vaccine improves LRT-mediated survival of mice with CT26 tumors in a CD8+ T cell-dependent manner. (a–c) CT26 tumor growth (a) and survival (b) of BALB/c mice (n = 7–8/group) locally irradiated with 12 Gy or 3 × 6 Gy at a mean volume of 60 mm3. (c) gp70-AH1 tetramer+ CD8+ T cells in blood of treated mice (n = 4–5/group). (d-f) CT26 tumor growth (d) and survival (e) of mice (n = 7–12/group) locally irradiated with 12 Gy at a mean tumor volume of ~70 mm3 and immunized three times with CT26 PME1 or control RNA-LPX. (f) Gp70-AH1 tetramer+ CD8+ T cells in blood of treated mice (n = all mice/group). (g-i) CT26 tumor growth (g) and survival (h) of mice (n = 8–9/group) immunized with CT26 PME1 or control RNA and locally irradiated at a mean tumor volume of 90 mm3. CD8+ T cells were depleted 6 days after LRT, administering the anti-CD8 antibody every 3–4 days over 3 weeks. (i) Gp70-AH1 tetramer+ CD8+ T cells in blood of treated mice (n = 8–9/group). Significance was determined using (b, e, h) Mantel-cox log-rank test and (c, f, i) one-way ANOVA, Tukey’s multiple comparison test. (a, d, g) Tumor growth is displayed on a log2-scale. Ratios depict frequency of mice with complete tumor responses (CR). Mean±SEM. nd = not determined.
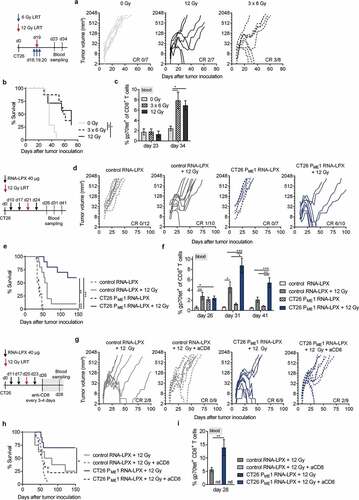
Figure 2. Activated poly-functional Th1-like CD4+ T cells against the immunodominant vaccine-encoded CD4 neoantigen are induced in the spleen of CD4 neoantigen vaccine/LRT treated mice. (a–c) CT26 tumor growth (a) and survival (b) of BALB/c mice (n = 7–10/group) treated with 12 Gy at a mean tumor volume of 45 mm3 and immunized with different RNA-LPX vaccines. RNA-LPX vaccines included CT26 PME1; CT26 ME1, the most immunogenic CT26 PME1 neoantigen; Pctrl. encoding mutations not expressed in CT26 and control RNA-LPX, encoding no antigens at all. (c) Gp70-AH1 tetramer+ CD8+ T cells in treated mice (n = 7–10/group). (d, e) Phenotypic analysis of enriched splenic CD4+ T cells from mice immunized with CT26 PME1 or control RNA-LPX (n = 5/group). (d) Differential expression of ICOS, CD69, PD-1, CD62L, T-bet, CD25 on ME1-specific (ME1tet+) and -nonspecific (ME1tet−) CD4+ T cells as determined by flow cytometry. Representative pseudocolor plots of ME1 tetramer staining are shown. (e) Supernatant cytokine secretion after 48 h co-culture of CD4+ T cells with ME1-peptide-loaded BALB/c BMDC. (f) IFNγ intracellular cytokine staining after ex vivo re-stimulation of enriched splenic CD4+ T cells from CT26-tumor-bearing mice, locally 12 Gy irradiated at a mean tumor volume of 60 mm3 and immunized with CT26 PME1 or control RNA-LPX, with ME1 peptide-loaded BMDC (n = 6/group). Significance was determined using (b) Mantel-Cox log-rank test, (c, d, f) one-way ANOVA, Tukey’s multiple comparison test and (e) unpaired, two-tailed Student’s t-test. (a) Tumor growth is displayed on a log2-scale. Ratios depict frequency of mice with complete tumor responses (CR). Mean±SEM.
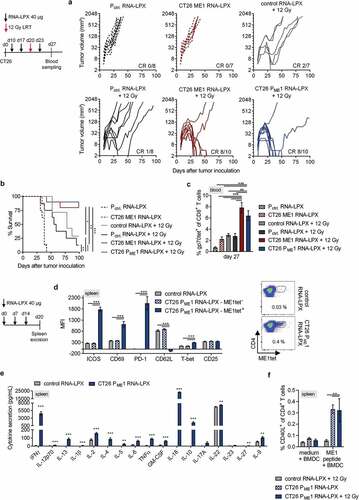
Figure 3. Adding the CD4 neoantigen vaccine to LRT results in a potent polyantigenic CD8+ T cell response and T cell memory. (a) IFNγ ELISpot using splenocytes isolated from CT26 tumor-bearing BALB/c mice, vaccinated with CT26 PME1 or control RNA-LPX and locally irradiated at a mean tumor volume of 45 mm3 (n = 6 mice/group, 2 mice pooled each), against CT26 cells, gp70-AH1 peptide-pulsed BALB/c BMDC, CT26-gp70KO, and 20 Gy irradiated CT26-gp70KO cells. In vitro irradiation was performed to enhance tumor cell MHC class I presentation (Supplementary Figure 3(a)). (b-d) CT26-gp70KO tumor growth (b) and survival (c) of mice immunized with CT26 PME1 or control RNA-LPX and irradiated at a mean volume of 45 mm3 (n = 7–10/group). (d) IFNγ ELISpot using splenocytes isolated from CT26 tumor-bearing mice (n = 4 mice/group, 2 mice pooled each) against CT26 cells, 20 Gy irradiated CT26-gp70KO cells and gp70-AH1 peptide-pulsed BALB/c BMDC. As in (a), in vitro irradiation was performed to enhance tumor cell MHC class I presentation (Supplementary Figure 3(a)). (e) Survival of 12 Gy and CT26 PME1/ME1 RNA-LPX treated CT26 tumor-free mice, challenged with a tumorigenic dose of CT26-gp70KO cells 40 days after initial tumor rejection (n = 10 each). Naïve BALB/c mice served as control group (n = 10). Significance was determined using (c, e) log-rank test and (a, d) one-way ANOVA, Tukey’s multiple comparison test. (b) Tumor growth is displayed on a log2-scale. Ratios depict frequency of mice with complete tumor responses (CR). Mean±SEM.
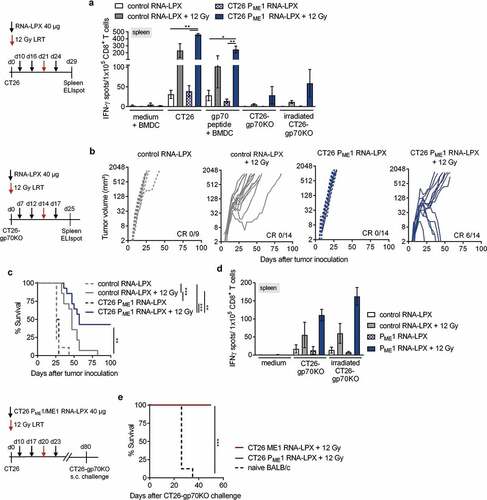
Figure 4. Tumor immune infiltrates of CD4 neoantigen vaccine/LRT-treated mice consist of activated, tumor antigen-specific CD8+ T cells and vaccine-induced CD4+ T cells capable of providing cognate help. (a, b) Analysis of TIL in CT26 tumor-bearing BALB/c mice (n = 5–6/group) treated with 12 Gy at a mean tumor volume of 45 mm3 and immunized with CT26 PME1 or control RNA-LPX. Tumors were resected 8 days after LRT. (a) Percentage of CD45+, tumor cells, CD4+ and CD8+ T cells and (b) gp70-AH1 tetramer+ CD8+ T cells. Representative pseudocolor plots show gp70-AH1 tetramer staining. (c-e) Analysis of tumor-infiltrating CD8+ T cells (c, d) and CD4+ T cells (e) from CT26 tumor-bearing mice locally irradiated with 12 Gy at a mean tumor volume of 60 mm3 and immunized three times with CT26 PME1 or control RNA-LPX (n = 6/group). CD4+ and CD8+ T cells were enriched via CD45+ TIL MACS for (c) phenotypic analysis or (d, e) intracellular cytokine staining after restimulation with gp70-AH1 or ME1 peptide-loaded BALB/c BMDC. Representative pseudocolor plots show CD8+ (d, right) and CD4+ (e, right) T cells after peptide restimulation. (f, g) TCRβ CDR3 sequencing of tumor CD8+ T cells from mice (n = 3–4/group), irradiated with 12 Gy at a mean tumor volume of ~100 mm3 and immunized three times with CT26 PME1 RNA-LPX or control RNA-LPX, analyzed 9 days after irradiation. (g) Number of unique TCRβ sequences in tumors or treated mice (left) and mean TCRβ clonotype frequency (clonality) of CD8+ T cells (right). (f) Frequency of blood-tumor shared TCRβ clones prior and 9 days after LRT compared to all unique tumor clones found 9 days after LRT. Significance was determined using (a–g) one-way ANOVA, Tukey’s multiple comparison test. Mean±SEM.
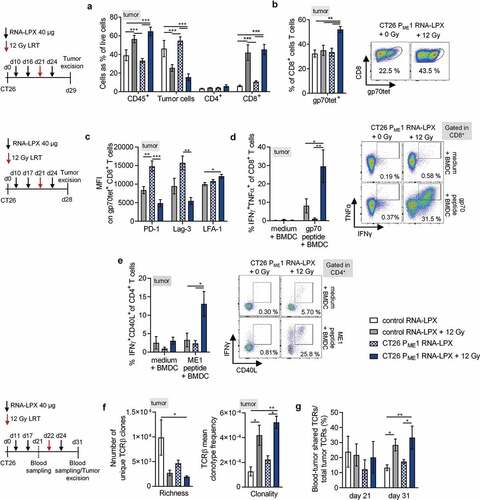
Figure 5. CD4 neoantigen vaccine/LRT treatment followed by anti-CTLA4 antibody therapy further enhances the efficacy with complete remission of gp70-negative CT26 tumors and survival of all mice. (a–c) CT26-gp70KO tumor growth (a) and survival (b) of BALB/c mice (n = 13–14/group) locally irradiated with 12 Gy at a mean tumor volume of 60 mm3, immunized three times with CT26 ME1 or control RNA-LPX and treated with anti-CTLA-4 or anti-PD-1 antibodies 3, 8, and 13 days after LRT. (c) IFNγ ELISpot using peripheral blood lymphocytes against wild-type or irradiated CT26-gp70KO cells (n = 13–14/group, blood of 3–4 mice pooled/data point). Significance was determined using (b) Mantel-Cox log-rank test and (c) one-way ANOVA, Tukey’s multiple comparison test. (a) Tumor growth is displayed on a log2-scale. Ratios depict frequency of mice with complete tumor responses (CR). Mean±SEM.
Author contributions
U.S., F.V., S.K., M.D., M.V. and J.Q. conceived and guided the study. N.S. and A.S. performed experiments. N.S. analyzed experiments. B.S., M.L., J.Q. and T.B. analyzed sequencing data. N.S., Ö.T., U.S. and S.K. interpreted the results and wrote the manuscript.
Disclosure of potential conflicts of interest
M.V., J.Q., S.K., M.D., Ö.T., and U.S. are employees at BioNTech (Mainz, Germany). U.S., M.D., M.V., M.L., B.S. and S.K. are inventors on patents and patent applications related to this study. U.S. is cofounder; Ö.T. and U.S management board member of BioNTech and have an ownership interest in TRON and BioNTech. All other authors have no potential conflict of interest.
{kind=link}
Acknowledgments
We thank I. Beulshausen, M. Brkic, E. Daniel, E. Petscherskich, A. Olbermann, E. Ockfen, H. Junginger and L. Cao for technical assistance; S. Witzel and B. Tillmann for cloning of constructs; E. Böhm, R. Sinderwald, F. Fleckenstein and S. Fesser for RNA production; V. Bukur, J. de Graf and C. Albrecht for the next-generation sequencing; H.Hefesha, J. Schumacher, J. Erk, A. Gerdts for liposomal formulation and RNA-LPX measurement. We are grateful to L.M. Kranz, C. Hotz and J. Beck for conceptual and technical discussions. We are grateful for the production of MHC-tetramers by the NIH Tetramer Core Facility.
Supplementary
Supplemental data for this article can be accessed on the publisher’s website.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Orth M, Lauber K, Niyazi M, et al. Current concepts in clinical radiation oncology. Radiat Environ Biophys. 2014;53(1):1–29. doi:10.1007/s00411-013-0497-2.
- Gupta A, Probst HC, Vuong V, Landshammer A, Muth S, Yagita H, Schwendener R, Pruschy M, Knuth A, van den Broek M, et al. Radiotherapy promotes tumor-specific effector cd8+t cells via dendritic cell activation. The Journal of Immunology. 2012;189(2):558–13. (Baltimore, Md.: 1950). doi:10.4049/jimmunol.1200563.
- Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nature Reviews Immunology. 2017;17(2):97–111. doi:10.1038/nri.2016.107.
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li X-D, Mauceri H, Beckett M, Darga T, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type i interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41(5):843–852. doi:10.1016/j.immuni.2014.10.019.
- Formenti SC, Demaria S. Radiation therapy to convert the tumor into an in situ vaccine. Int J Radiat Oncol Biol Phys. 2012;84(4):879–880. doi:10.1016/j.ijrobp.2012.06.020.
- Formenti SC, Rudqvist N-P, Golden E, Cooper B, Wennerberg E, Lhuillier C, Vanpouille-Box C, Friedman K, Ferrari de Andrade L, Wucherpfennig KW, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med. 2018;24(12):1845–1851. doi:10.1038/s41591-018-0232-2.
- Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu Y-X. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014;124(2):687–695. doi:10.1172/JCI67313.
- Honeychurch J, Glennie MJ, Johnson PWM, Illidge TM. Anti-CD40 monoclonal antibody therapy in combination with irradiation results in a CD8 T-cell-dependent immunity to B-cell lymphoma. Blood. 2003;102(4):1449–1457. doi:10.1182/blood-2002-12-3717.
- Rodriguez-Ruiz ME, Rodriguez I, Garasa S, Barbes B, Solorzano JL, Perez-Gracia JL, Labiano S, Sanmamed MF, Azpilikueta A, Bolanos E, et al. Abscopal effects of radiotherapy are enhanced by combined immunostimulatory mabs and are dependent on Cd8 T cells and crosspriming. Cancer Res. 2016;76(20):5994–6005. doi:10.1158/0008-5472.CAN-16-0549.
- Zheng W, Skowron KB, Namm JP, Burnette B, Fernandez C, Arina A, Liang H, Spiotto MT, Posner MC, Fu Y-X, et al. Combination of radiotherapy and vaccination overcomes checkpoint blockade resistance. Oncotarget. 2016;7(28):43039–43051. doi:10.18632/oncotarget.9915.
- Mondini M, Nizard M, Tran T, Mauge L, Loi M, Clemenson C, Dugue D, Maroun P, Louvet E, Adam J, et al. Synergy of Radiotherapy and a Cancer Vaccine for the Treatment of HPV-Associated Head and Neck Cancer. Mol Cancer Ther. 2015;14(6):1336–1345. doi:10.1158/1535-7163.MCT-14-1015.
- Witek M, Blomain ES, Magee MS, Xiang B, Waldman SA, Snook AE. Tumor radiation therapy creates therapeutic vaccine responses to the colorectal cancer antigen GUCY2C. Int J Radiat Oncol Biol Phys. 2014;88(5):1188–1195. doi:10.1016/j.ijrobp.2013.12.043.
- Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520(7549):692–696. doi:10.1038/nature14426.
- Slansky JE, Rattis FM, Boyd LF, Fahmy T, Jaffee EM, Schneck JP, Margulies DH, Pardoll DM. Enhanced antigen-specific antitumor immunity with altered peptide ligands that stabilize the MHC-peptide-TCR complex. Immunity. 2000;13(4):529–538. doi:10.1016/S1074-7613(00)00052-2.
- Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534(7607):396–401. doi:10.1038/nature18300.
- Orlandini von Niessen AG, Poleganov MA, Rechner C, Plaschke A, Kranz LM, Fesser S, Diken M, Löwer M, Vallazza B, Beissert T, et al. Improving mRNA-based therapeutic gene delivery by expression-augmenting 3ʹ UTRs identified by cellular library screening. Mol Ther: J Am Soc Gene Ther. 2019;27(4):824–836. doi:10.1016/j.ymthe.2018.12.011.
- Holtkamp S, Kreiter S, Selmi A, Simon P, Koslowski M, Huber C, Türeci O, Sahin U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. 2006;108(13):4009–4017. doi:10.1182/blood-2006-04-015024.
- Kuhn AN, Diken M, Kreiter S, Selmi A, Kowalska J, Jemielity J, Darzynkiewicz E, Huber C, Türeci Ö, Sahin U, et al. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 2010;17(8):961–971. doi:10.1038/gt.2010.52.
- Kreiter S, Selmi A, Diken M, Sebastian M, Osterloh P, Schild H, Huber C, Türeci Ö, Sahin U. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. The Journal of Immunology. 2008 (Baltimore, Md.: 1950);180(1):309–318. doi:10.4049/jimmunol.180.1.309.
- Grunwitz C, Salomon N, Vascotto F, Selmi A, Bukur T, Diken M, Kreiter S, Türeci Ö, Sahin U. HPV16 RNA-LPX vaccine mediates complete regression of aggressively growing HPV-positive mouse tumors and establishes protective T cell memory. Oncoimmunology. 2019;8(9):e1629259. doi:10.1080/2162402X.2019.1629259.
- Sahin U. et al. An RNA vaccine drives immunity in checkpoint inhibitor-experienced melanoma. Nature , 2020, accepted
- Heesen L, Jabulowsky R, Loquai C, Utikal J, Gebhardt C, Hassel J, Kaufmann R, Pinter A, Derhovanessian E, Diken M, et al. 49P A first-in-human phase I/II clinical trial assessing novel mRNA-lipoplex nanoparticles encoding shared tumor antigens for potent melanoma immunotherapy. Annals of Oncology. 2017;28(mdx711):030. doi:10.1093/annonc/mdx711.030.
- Jabulowsky RA, Loquai C, Derhovanessian E, Mitzel-Rink H, Utikal J, Hassel J, Kaufmann R, Pinter A, Diken M, Gold M, et al. 1238TiPA first-in-human phase I/II clinical trial assessing novel mRNA-lipoplex nanoparticles encoding shared tumor antigens for immunotherapy of malignant melanoma. Annals of Oncology. 2018;29:viii439. doi:10.1093/annonc/mdy288.109.
- Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–226. doi:10.1038/nature23003.
- Huang AY, Gulden PH, Woods AS, Thomas MC, Tong CD, Wang W, Engelhard VH, Pasternack G, Cotter R, Hunt D, et al. The immunodominant major histocompatibility complex class I-restricted antigen of a murine colon tumor derives from an endogenous retroviral gene product. Proc Natl Acad Sci U S A. 1996;93(18):9730–9735. doi:10.1073/pnas.93.18.9730.
- Vormehr M, Reinhard K, Blatnik R, Josef K, Beck JD, Salomon N, Suchan M, Selmi A, Vascotto F, Zerweck J, et al. A non-functional neoepitope specific CD8 + T-cell response induced by tumor derived antigen exposure in vivo. Oncoimmunology. 2019;8(3):1553478. doi:10.1080/2162402X.2018.1553478.
- Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka. Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinf (Oxford, England). 2012;28(14):1811–1817. doi:10.1093/bioinformatics/bts271.
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–576. doi:10.1101/gr.129684.111.
- Kreiter S, Konrad T, Sester M, Huber C, Türeci Ö, Sahin U. Simultaneous ex vivo quantification of antigen-specific CD4+ and CD8+ T cell responses using in vitro transcribed RNA. Cancer Immunol Immunother: CII. 2007;56(10):1577–1587. doi:10.1007/s00262-007-0302-7.
- Gough MJ, Crittenden MR, Young KH. Comparing equals when evaluating immunotherapy with different doses and fractions of radiation therapy. Immunother. 2015;7(8):847–849. doi:10.2217/IMT.15.58.
- Diken M, Vormehr M, Grunwitz C, Kreiter S, Türeci Ö, Sahin U. Discovery and Subtyping of Neo-Epitope Specific T-Cell Responses for Cancer Immunotherapy: Addressing the Mutanome. Methods Mol Biol. 2017;1499:223–236. doi:10.1007/978-1-4939-6481-9_14.
- Chattopadhyay PK, Yu J, Roederer M. A live-cell assay to detect antigen-specific CD4+ T cells with diverse cytokine profiles. Nat Med. 2005;11(10):1113–1117. doi:10.1038/nm1293.
- Kreiter S, Selmi A, Diken M, Koslowski M, Britten CM, Huber C, Tureci O, Sahin U. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 2010;70(22):9031–9040. doi:10.1158/0008-5472.CAN-10-0699.
- Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, Chudakov DM. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods. 2015;12(5):380–381. doi:10.1038/nmeth.3364.
- Shugay M, Bagaev DV, Turchaninova MA, Bolotin DA, Britanova OV, Putintseva EV, Pogorelyy MV, Nazarov VI, Zvyagin IV, Kirgizova VI, et al. VDJtools: unifying post-analysis of T cell receptor repertoires. PLoS Comput Biol. 2015;11(11):e1004503. doi:10.1371/journal.pcbi.1004503.
- Kim H-J, Cantor H. CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol Res. 2014;2(2):91–98. doi:10.1158/2326-6066.CIR-13-0216.
- Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, K.Wansley E, Camphausen K, Luiten RM, de Ru AH, Neijssen J, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203(5):1259–1271. doi:10.1084/jem.20052494.
- Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520(7547):373–377. doi:10.1038/nature14292.
- Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. Using immunotherapy to boost the abscopal effect. Nature Reviews Cancer. 2018;18(5):313–322. doi:10.1038/nrc.2018.6.
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548(7668):466–470. doi:10.1038/nature23470.
- Brix N, Tiefenthaller A, Anders H, Belka C, Lauber K. Abscopal, immunological effects of radiotherapy: narrowing the gap between clinical and preclinical experiences. Immunol Rev. 2017;280(1):249–279. doi:10.1111/imr.12573.
- Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, Beckett M, Sharma R, Chin R, Tu T, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114(3):589–595. doi:10.1182/blood-2009-02-206870.
- Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. The Journal of Immunology. 2005 (Baltimore, Md.: 1950);174(12):7516–7523. doi:10.4049/jimmunol.174.12.7516.
- Dovedi SJ, Cheadle EJ, Popple AL, Poon E, Morrow M, Stewart R, Yusko EC, Sanders CM, Vignali M, Emerson RO, et al. Fractionated radiation therapy stimulates antitumor immunity mediated by both resident and infiltrating polyclonal T-cell populations when combined with PD-1 blockade. Clin Cancer Res: Off J Am Assoc Cancer Res. 2017;23(18):5514–5526. doi:10.1158/1078-0432.CCR-16-1673.
- Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, Demaria S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res: Off J Am Assoc Cancer Res. 2009;15(17):5379–5388. doi:10.1158/1078-0432.CCR-09-0265.
- Chakraborty M, Abrams SI, Coleman CN, Camphausen K, Schlom J, Hodge JW. External beam radiation of tumors alters phenotype of tumor cells to render them susceptible to vaccine-mediated T-cell killing. Cancer Res. 2004;64(12):4328–4337. doi:10.1158/0008-5472.CAN-04-0073.
- Fotin-Mleczek M, Zanzinger K, Heidenreich R, Lorenz C, Kowalczyk A, Kallen K-J, Huber SM. mRNA-based vaccines synergize with radiation therapy to eradicate established tumors. Radiat Oncol (London, England). 2014;9(1):180. doi:10.1186/1748-717X-9-180.
- Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4+ T cell help in cancer immunology and immunotherapy. Nature Reviews Immunology. 2018;18(10):635–647. doi:10.1038/s41577-018-0044-0.
- Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207(3):637–650. doi:10.1084/jem.20091918.
- Alam S, Knowlden ZAG, Sangster MY, Sant AJ. CD4 T cell help is limiting and selective during the primary B cell response to influenza virus infection. J Virol. 2014;88(1):314–324. doi:10.1128/JVI.02077-13.
- Ahrends T, Spanjaard A, Pilzecker B, Bąbała N, Bovens A, Xiao Y, Jacobs H, Borst J. CD4+ T cell help confers a cytotoxic T cell effector program including coinhibitory receptor downregulation and increased tissue invasiveness. Immunity. 2017;47(5):848–861.e5. doi:10.1016/j.immuni.2017.10.009.
- Vasiliou V, Nebert DW. Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. Hum Genomics. 2005;2(2):138–143. doi:10.1186/1479-7364-2-2-138.
- Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, Boisguerin V, Bukur T, Sorn P, Paret C, et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics. 2014;15(1):190. doi:10.1186/1471-2164-15-190.
- Dovedi SJ, Adlard AL, Lipowska-Bhalla G, McKenna C, Jones S, Cheadle EJ, Stratford IJ, Poon E, Morrow M, Stewart R, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014;74(19):5458–5468. doi:10.1158/0008-5472.CAN-14-1258.
- Wang X, Schoenhals JE, Li A, Valdecanas DR, Ye H, Zang F, Tang C, Tang M, Liu C-G, Liu X, et al. Suppression of Type I IFN signaling in tumors mediates resistance to anti-PD-1 treatment that can be overcome by radiotherapy. Cancer Res. 2017;77(4):839–850. doi:10.1158/0008-5472.CAN-15-3142.
- Takeshima T, Chamoto K, Wakita D, Ohkuri T, Togashi Y, Shirato H, Kitamura H, Nishimura T. Local radiation therapy inhibits tumor growth through the generation of tumor-specific CTL: its potentiation by combination with Th1 cell therapy. Cancer Res. 2010;70(7):2697–2706. doi:10.1158/0008-5472.CAN-09-2982.
- Goding SR, Wilson KA, Antony PA. Combination of adoptive cell transfer, anti-PD-L1 and anti-LAG-3 antibodies for the treatment of recurrent tumors. Oncoimmunology. 2014;2(8):e25050. doi:10.4161/onci.25050.
- Vormehr M, Türeci Ö, Sahin U. Harnessing tumor mutations for truly individualized cancer vaccines. Annu Rev Med. 2019;70(1):395–407. doi:10.1146/annurev-med-042617-101816.
- Schumacher TN, Scheper W, Kvistborg P. Cancer neoantigens. Annu Rev Immunol. 2019;37(1):173–200. doi:10.1146/annurev-immunol-042617-053402.