
ABSTRACT
Adoptive T cell therapy has proven effective against hematologic malignancies and demonstrated efficacy against a variety of solid tumors in preclinical studies and clinical trials. Nonetheless, antitumor responses against solid tumors remain modest, highlighting the need to enhance the effectiveness of this therapy. Genetic modification of T cells with RNA has been explored to enhance T-cell antigen specificity, effector function, and migration to tumor sites, thereby potentiating antitumor immunity. This review describes the rationale for RNA-electroporated T cell modifications and provides an overview of their applications in preclinical and clinical investigations for the treatment of hematologic malignancies and solid tumors.
Introduction
Cancer immunotherapy is a promising approach for potentiating antitumor immunity, with the ability to promote long-lasting antitumor immune responses while restricting the toxicities caused by conventional cancer treatments such as radiation and chemotherapy. Consequently, a number of immunotherapeutic strategies have been evaluated to target cancer over the past couple of decades. Such strategies have included an abundance of vaccine‐based strategies to trigger cellular and humoral anti‐tumor immunity, as well as antibody‐based strategies to mediate complement and natural killer cell (NK)‐dependent anti‐tumor cytolytic activity, block inhibitory receptors, or modulate the tumor microenvironment.Citation1–Citation5 However, these approaches are limited by central and peripheral immune tolerance, which prevent immune effector cells from effectively targeting tumor cells due to the lack of a T‐cell repertoire to self‐antigens overexpressed or aberrantly expressed by tumors. The establishment of an immune‐suppressive state also constitutes a limitation to cancer immunotherapies. The use of immune checkpoint blocking antibodies, such as anti-PD1, has proven to be a successful and safe strategy for overcoming peripheral tolerance and has been shown to abrogate tumor growth and enhance antitumor immune responses in multiple preclinical as well as clinical trial studies.Citation6–Citation10 As a result, immune checkpoint inhibitors have recently been FDA approved for the treatment of various cancers.
One approach to overcome central tolerance has been the transfer of potent tumor-specific effector cells into patients through adoptive cell therapy (ACT). The rationale for ACT of T cells stems from the central role of T lymphocytes in tumor antigen recognition and cell-mediated immunity. Unlike other immunotherapy modalities that rely on endogenous tumor responses, ACT is based on the isolation of T cells from the patient’s tumor (tumor-infiltrating lymphocytes) or peripheral blood, ex vivo expansion of tumor-specific unmodified or genetically engineered lymphocytes, and infusion of these T cells back into the patient.Citation11,Citation12 The feasibility and efficacy of ACT have been clearly documented in melanoma patients and a few other solid tumors, such as ovarian and colorectal cancers, as well as some B cell malignancies.Citation12–Citation19 Although numerous preclinical studies have demonstrated the potential value of ACT for various cancers, only a few of them have been successfully translated into the clinic. Major hurdles to the success ACT are summarized in . Improvements in this potentially powerful treatment intervention are therefore needed.
Figure 1. Challenges in Adoptive Cell Therapy. Illustrative figure describing the major limitations of the adoptive T cell transfer in different categories including antigen-specificity, effector T cell function and T cell persistence and migration. (ACT) adoptive cell transfer.
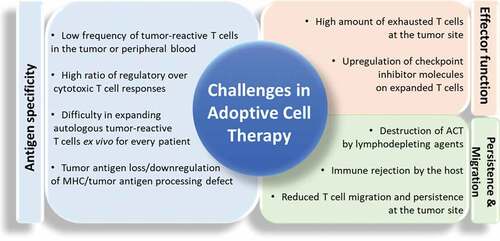
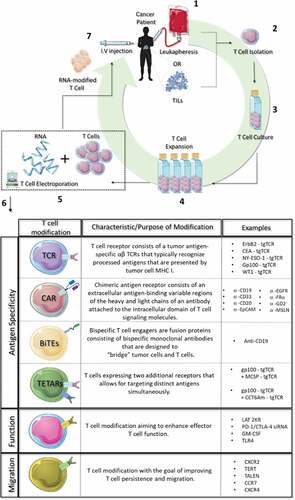
Figure 2. RNA-electroporated T cells. 1. Peripheral blood cells (leukapheresis) or tumor infiltrating lymphocytes (TILs) are collected from cancer patients. 2. T cells are isolated from blood or TILs by density gradient centrifugation. 3. Isolated T cells are incubated at 37οC. 4. T cells are expanded in vitro with cytokines (e.g.IL-2). 5. T cells are combined with RNA and transfected with an electroporation device. 6. Types of RNA T cell modifications. 7. RNA-modified T cells are injected back into the same patient.
The advancements made in tumor antigen discovery and ex vivo culture of T cells have led to the development of strategies aimed at augmenting antitumor responses through the reprogramming of T cells prior to ACT.Citation20–Citation42 Methods for T cell modification have included the use of viral vectors (e.g., retroviruses and lentiviruses) and non-viral vectors (e.g., electroporation (EP) and liposomes) for DNA or RNA delivery. Each of those methods presents its own advantages and disadvantages (as summarized in ), which have previously been reviewed in detail by others.Citation43–Citation47 Traditionally, T cell modifications have been achieved through DNA-mediated, viral vector platforms.Citation48,Citation49 However, while preclinical and clinical studies have demonstrated the antitumor potency of virally engineered T cells, these studies have also highlighted substantial regulatory hurdles (e.g., clinical production of the plasmids, as they are associated with genes for antibiotic resistance; removal of viable residual packaging cells). These hurdles hinder implementation in human clinical trials and adoption into clinical practice due to the toxicities associated with long-term transgene expression or the potential presence of endotoxin in the viral vector preparation. Therefore, significant preclinical and clinical research have been dedicated to developing alternative vector systems that are not dependent on viral design, such as mRNA transfer that has proven to be safe and efficacious in both preclinical and clinical studies.Citation20,Citation23,Citation25,Citation26
Table 1. Comparison of advantages and limitations of viral vectors versus RNA transfection for T cell modification.
To date, RNA-mediated modifications of T cells have been aimed at enhancing antigenic specificity, effector function, persistence, and/or migration to tumor sites, as summarized in .Citation21–Citation26,Citation28-Citation35,Citation37–Citation42,Citation50-Citation55 The modifications made to enhance T-cell antigenic specificity include addition of a tumor antigen-specific T cell receptor (TCR), chimeric antigen receptor (CAR), bispecific T cell engagers (BiTEs), and T cell expressing two additional receptors (TETARs). Recently, RNA-modified T cells have also been explored as biological carriers to intracranial tumorsCitation52 as well as an effective tool for immunomodulatory agent delivery for cancer therapy.Citation56 The present review provides an overview of various T cell modifications achieved through RNA electroporation and their progress in the treatment of hematologic malignancies and solid tumors.
Improvement of T-cell antigen specificity
Effective T cell-based therapies require a directed and synchronized process in order to generate an adaptive cell-mediated immune response. In order to mount an effective immune response, T cells are initially primed by tumor peptides presented by major histocompatibility complexes (MHCs) on antigen-presenting cells (APCs) in the lymph nodes. T cells recognize tumor-associated antigens or tumor-specific antigens through the TCR, a transmembrane complex composed of two subunits (α and β chains).Citation57 This recognition leads to the activation and migration of T cells to the tumor, consequently eliciting cytotoxic T cell functions.Citation57 However, most tumors are poorly immunogenic,Citation58–Citation61 and the peripheral T cell repertoire typically anergic,Citation62 devoid of high-avidity tumor-reactive T cellsCitation63 and favoring the differentiation of regulatory over cytotoxic T cell responses.Citation64 Therefore, preclinical research has focused on developing methods to reprogram T cells ex vivo so they can express a diverse repertoire of tumor-specific antigen recognition receptors such as TCRs or CARs. While tumor-specific αβ TCRs typically recognize processed antigens that are presented by tumor cells’ MHC Class I, CARs have been designed to overcome this MHC-restricted recognition through targeting antigenic peptides expressed at the cell surface.Citation46
T cells engineered to express tumor-specific TCRs or CARs using viral vectors have shown promising clinical outcomes in ACT for various cancers.Citation48,Citation49,Citation65 However, this strategy has several limitations, such as “on-target” or “off-target” effects that can be potentiated by long-term transgene expression.Citation45–Citation47 Therefore, methods exploring non-viral, RNA-mediated modifications of T cells have been investigated as potential, similarly efficacious, and safer alternatives to stable expression via viral vectors.Citation66 RNA EP has been used to enhance T-cell antigen specificity, mainly through the expression of tumor-specific TCRs, CARs, TETARs, or BiTEs.Citation26,Citation32–Citation34,Citation67
TCRs
EP of mRNA into immune cells was first employed to modify dendritic cells (DCs) so they express tumor antigens or to functionally modulate their phenotype.Citation68–Citation70 Subsequently, mRNA EP has been used to transfer full-length, tumor-specific TCR α and β chain genes into T cells in order to redirect their antigen specificity.Citation31,Citation33 The impetus for reprogramming T cells’ TCR mainly stemmed from the challenges in isolating and expanding autologous tumor-reactive T cells ex vivo for every patient receiving ACT. Additionally, the low frequency of high-affinity, antigen-specific CD8+ T cells among cancer patients’ peripheral blood lymphocytes (PBLs) is a limiting factor in tumor cell killing efficiency.Citation71
Zhao et al. were the first to report T cell EP with TCR RNA in human primary PBLs.Citation32 In this study, TCR RNA was isolated from activated PBLs stimulated with New York esophageal squamous cell cancer-1 (NY-ESO-1)-specific peptide, and TCR RNA EP was used to screen for TCR functionality prior to generating viral vector constructs. Co-culture of NY-ESO-1 TCR RNA-modified T cells with tumor cells resulted in the killing of cognate human tumor cells, thus demonstrating functional human leukocyte antigen (HLA)-A2-restricted and NY-ESO-1-specific TCR α- and β-chains.Citation32
Since 2005, a handful of preclinical studies using TCR RNA-electroporated T cells against solid tumors have been reported for different types of cancer (e.g., neuroblastoma, ovarian cancer, melanoma), as shown in . These reports have demonstrated that transfecting lymphocytes with tumor-specific TCR RNA can redirect CD8+ T cells to lyse targeted tumor cells specifically and elicit similar cytotoxic capacity as observed with retrovirally transduced T cells.Citation29,Citation31,Citation33
Table 2. Representative preclinical studies using RNA-based modifications of T cells for solid tumors. CAR, chimeric antigen receptor; CCT6Am, mutation of chaperonin containing TCP1, subunit 6A; CSPG4, chondroitin sulfate proteoglycan 4; EpCAM, epithelial cell adhesion molecule CD326; MCSP, melanoma-associated chondroitin sulfate proteoglycan; TCR, T cell receptor; TETARs, T cell expressing two additional receptors.
In addition, T cells modified with RNA can be used to screen highly antigen–binding regions of TCRs in order to modulate and enhance tumor cell recognition by CD8+ and CD4+ T cells. In transgenic mice expressing fully human HLA-A2.1 (previously immunized with HLA-A2.1-restricted carcinoembryonic antigen (CEA) epitopes), RNAs encoding for amino acid substitutions of the TCR α- and β-chains were transfected into CD8+ and CD4+ T cells. While replacement of a single amino acid of the TCR enhanced tumor cell recognition by CD8+ T cells, dually replaced TCR amino acid positions enhanced tumor cell recognition by CD4+ T cells. In response to HLA-A2.1 and CEA-expressing human colon cancer cell lines, or genetically engineered cell lines, these TCR modifications led to functional effector T cell functions as detected by IFNγ secretion.Citation28
A potential drawback to T cell TCR reprogramming is the dimer formation that can occur between endogenous and transferred TCR chains, which has shown to affect T cell specificity.Citation72 To overcome this challenge, Shimizu et al. evaluated the antitumor effects of γδ T cells that were modified to express an invariant natural killer T cell (iNKT) TCR (an innate lymphocyte’s TCR).Citation37 Since γδ T cells lack α- and β-chains, the authors hypothesized that transfection of iNKT α- and β-chain mRNA into γδ T cells would prevent the formation of mispaired receptors. iNKT-TCR mRNA-transfected γδ T cells retained parental effector T cell functions and responded to both γδ T and glycolipid iNKT ligands; moreover, they showed cytotoxic T cell responses against iNKT ligand-expressing target cells and leukemia cells in vitro.Citation37 Together, these studies demonstrate that TCR RNA transfer can redirect and enhance T cell antigen specificity for tumor adoptive immunotherapy.
CARs
CARs consist of the extracellular antigen-binding, variable regions of the heavy and light chains of antibodies (specific to a particular antigen), a transmembrane domain, and a signaling endodomain. The addition of co-stimulatory signaling molecules can further enhance the antitumor immune responses and in vivo persistence of CAR T cells.Citation49 Importantly, CAR T cells recognize and kill tumor cells independently of MHC. This is a relevant characteristic, as many tumors present immune evasion mechanisms through which antigen processing and presentation are affected.Citation73 However, this feature also limits CARs to the exclusive targeting of membrane-bound antigens. A remarkable milestone in cancer immunotherapy has been evidenced by the FDA approval of an anti-CD19 autologous CAR T cell therapy for the treatment of leukemia patients who have been refractory to standard treatments or later relapsed. Although CAR T cell therapy has shown success in treating those patients, this treatment modality can also induce autoimmune responses and cytokine release syndrome, Citation74 which have in some cases been associated with lethal toxicity. These effects have been attributed to “off-target” tissue toxicity, which correlated with prolonged transgene expression in CAR T cells generated using viral vectors.Citation45,Citation46,Citation67 These toxicity concerns have prompted researchers to investigate the use of CAR RNA-modified T cells. A list of representative preclinical studies using RNA-based modifications of T cells for hematologic malignancies and solid tumors is shown in and .
Table 3. Representative preclinical studies using RNA-based modifications of T cells for hematological malignancies. α-GalCer, α-galactosylceramide; AML, acute myeloid leukemia; BiTE, bispecific T cell engager; CAR, chimeric antigen receptor; iNKT, invariant natural killer T; LAT, linker for activation of T cell; LAT 2KR, ubiquitylation-resistant LAT; TALEN, transcription activator-like effector nuclease; TERT, telomerase reverse transcriptase; Zol, zolendronic acid.
Hematologic malignancies
The successful application of CAR RNA-modified T cells in targeting B-cell antigens (e.g., CD19, CD20, and CD33) has been demonstrated in animal models of leukemia and lymphoma for hematologic malignancies.Citation21,Citation35,Citation38,Citation39,Citation41 Rabinovich et al. have shown the feasibility of generating CAR RNA-modified T cells following a simple and effective method, consisting of a vector-free tool production of mRNA from polymerase chain reaction-generated DNA templates, and tested this approach by generating CD19 CAR RNA-modified T cells that specifically targeted and lysed CD19+ donor matched cells.Citation42
Using bioluminescence imaging, studies have shown CAR RNA-modified T cell migration to tumor sites in mice with advanced leukemia. Following a single systemic anti-CD19 CAR RNA-transfected T cell injection in a leukemia xenograft model, these cells demonstrated efficient T cell persistence (at least 13 days post T cell infusion) and systemic migration in vivo.Citation40,Citation54 In another study, luciferase-expressing RNA-transfected CAR T cell distribution at sites of disease recurrence remained detectable at a time of expected loss of CAR expression.Citation40 Importantly, CAR RNA-transfected T cells exhibited similar antitumor efficacy as viral vector-modified T cells.Citation40
Other preclinical studies have used multiple CAR T cell infusions (equal doses), which resulted in tumor regression, Citation27,Citation50 but these studies have been largely performed in intraperitoneal/intratumoral models and without investigation of optimal treatment regimens. Barret et al. reported that weighted dose splitting (i.e., a loading dose followed by lower maintenance doses) of anti-CD19 RNA-transfected CAR T cells and interval lymphodepletion with cyclophosphamide (an alkylating agent used as chemotherapy) resulted in increased effectiveness and long-term durable remission (> 100 days) in a disseminated model of leukemia, when compared to mice that did not receive lymphodepletion.Citation40 In a xenograft mouse model of primary acute myeloid leukemia (AML), Kenderian et al. showed that RNA-transfected CAR T cells against CD33 (an antigen expressed on malignant and normal myeloid cells) elicited potent antitumor activity in vitro and resulted in leukemia eradication and long-term survival in vivo.Citation21
Promising outcomes of CAR T cell therapy in murine models of hematologic malignancies have led to the development of RNA-transfected CD20-specific CAR T cells for the treatment of dogs with spontaneous B cell lymphoma.Citation35 In this report, the authors demonstrated that canine CD20-ζ CAR was efficiently and transiently expressed in T cells after mRNA electroporation. Such treatment proved to be safe and efficacious, as demonstrated by effector T cell cytokine production and tumor cell killing in vitro, as well as modest and transient antitumor activity in a canine patient.
Solid tumors
The therapeutic potential of RNA-modified T cells has been demonstrated in many solid tumors, including mesothelioma, ovarian cancer, neuroblastoma, glioblastoma, and peritoneal carcinomatosis.Citation20,Citation24,Citation25,Citation27,Citation30,Citation50,Citation75 Unlike for blood-borne tumors, the success of RNA-transfected CAR T cells has not yet been extrapolated to solid cancers, mainly due to physical barriers impeding T cell tumor penetration, immunosuppressive tumor microenvironments, and limited T cell persistence at the tumor sites.Citation50,Citation76 In an initial study, Yoon et al. showed the therapeutic potential of CAR T cells generated by electroporation of PBLs with RNA encoding for anti-human epidermal growth factor receptor 2 (Her-2/neu) in a human ovarian carcinoma xenograft model.Citation30 Anti-Her-2/neu CAR T cells elicited potent immune responses against cognate antigen-overexpressing tumor cells in vitro and resulted in strong inhibition of tumor growth in vivo.Citation30 Similarly, Singh and colleagues demonstrated that a single intratumoral injection of autologous RNA-transfected CAR T cells directed against the neuroblastoma antigen disialoganglioside achieved antitumor effects and control of tumor growth in an orthotopic flank xenograft model of neuroblastoma.Citation75
In another study, Caruso et al. evaluated RNA-modified CAR T cells against epidermal growth factor receptor (EGFR), a TAA overexpressed in adult primary glioblastoma.Citation24 Following electroporation of human lymphocytes with EGFR-specific CAR RNA, T cells were exposed to cytokines or stimulated with EGFR-expressing parental mouse cells. While anti-EGFR CAR T cells specifically targeted tumor cells and secreted signature Th1 cytokines, the addition of cytokines in co-culture with cognate antigen-expressing tumor or normal cells accelerated transfected RNA decay, thereby reducing effector T cell function.Citation24 Together, these findings indicate that RNA-modified CAR T cells allow for “on-target” immune responses and the prevention of putative “off-target” toxicities that could be associated with viral vector-based T cell modifications.
A major concern regarding the clinical application of RNA-modified T cells has been the transient CAR expression on T cells due to physiologic decay over time, which could abrogate their capacity to target tumor cells and, therefore, reduce antitumor responses. To overcome this obstacle, studies have been applying multiple and weighted dosing strategies to avoid cancer relapse.Citation20,Citation25,Citation27,Citation75 For instance, in a preclinical model of mesothelioma, Zhao et al. tested RNA-transfected CAR T cells against mesothelin (MSLN), an antigen overexpressed in ovarian and pancreatic cancer as well as mesothelioma.Citation27 Sequential injections of autologous RNA-transfected MSLN CAR T cells promoted the dramatic regression of advanced flank and disseminated matched patient mesothelioma growth in vivo.Citation27 Moreover, phase I clinical trial employing mRNA-modified T cells for the treatment of MSLN-expressing tumors demonstrated the safety and feasibility of multiple injections of RNA-transfected MSLN-specific CAR T cells in most patients (NCT01355965).Citation20 A single patient was reported to have clinical anaphylaxis, which was most likely due to the generation of IgE antibodies specific to the murine CAR construct.Citation20 Together, these studies suggest that T cells engineered to express powerful activation domains through RNA electroporation represent an effective approach for adoptive cell transfer, without the associated safety concerns of integrating viral vectors.
TETARs
RNA-mediated T cell engineering has been mostly used to target a single TAA or TSA in order to improve antigen specificity. While this approach often leads to tumor regression, relapse is likely to occur due to: 1) immunoediting mechanisms triggered in the tumor microenvironment, 2) tumor antigen processing defect, 3) antigen loss, and/or 4) downregulation of MHC, Citation77,Citation78 among other factors. These tumor escape mechanisms highlight the therapeutic limitations likely to be encountered while targeting single antigens.
Recent investigations have developed the use of T cells expressing two additional receptors (TETARs) as a new therapeutic strategy to target two antigens simultaneously. Through combining two separate antigen receptors on the same T cell, this strategy has the potential to overcome immune escape due to single antigen loss. Hofflin et al. reported that activated human T cells could be effectively modified to express distinct TCRs directed against a common melanoma antigen and a patient-specific, individually mutated antigen. These dual-specific T cells specifically lysed target cells loaded with each of their cognate antigens in vitro, with some observed reciprocal inhibition.Citation26 In another study, Uslu et al. generated RNA-modified CD8+ TETARs that expressed both a CAR and TCR simultaneously, each receptor being specific for a distinct common melanoma antigen. Importantly, compared to viral vector-modified CAR and TCR T cells, these TETARs showed enhanced effector T cell functions without reciprocal inhibition.Citation23 Through functionally combining antigen-specific CARs and TCRs, RNA-modified TETARs could circumvent tumor immune escape, offering a promising alternative in T cell design for ACT therapy.
BiTEs
Bispecific T cell engagers (BiTEs) are fusion proteins consisting of bispecific monoclonal antibodies (mAbs) that are designed to “bridge” tumor cells and T cells.Citation79 This engagement facilitates the direct interaction between T cells and tumor antigens loaded on MHC. In xenograft animal models, BiTEs that have been stably transduced in human T cells have shown feasibility and efficacy, although permanent BiTE expression on T cells has the potential to elicit cytokine storms and promote B cell aplasia.Citation80,Citation81 To circumvent these potential toxicities, Liu et al. reported that BiTE T cells transfected with RNA encoding for an mAb targeting CD19 specifically secreted functional BiTEs and conferred specific tumor cell killing in vitro and improved overall survival in vivo.Citation34 Thus, BiTE RNA-modified T cells may represent an alternative to overcoming challenges of systemic BiTE injection while preventing toxicities resulting from current viral vector-mediated BiTE T cell therapies.
Improvement of T cell function
Effective antitumor T cell function involves a plethora of steps needed for the elimination of targeted cancer cells. T cells must first recognize tumor-specific antigens presented on APCs in the lymph nodes, upregulate activation markers and co-stimulatory molecules, proliferate extensively in vivo, and traffic to the tumor site while retaining their antitumor functions in a highly immunosuppressive tumor microenvironment. In physiological conditions, cytokines and chemokines tightly coordinate and regulate appropriate effector T cell functions. However, in cancer patients, tumor-reactive T cells are typically found at low frequency in the circulation and often fail to perform their inherent effector functions due to the upregulation of checkpoint inhibitor molecules (e.g., CTLA-4, PD-1) on their surface, downregulation of MHC Class I expression on tumor cells, secretion of anti-inflammatory molecules (e.g., TGFβ, IL-10), Citation77,Citation82–Citation85 and/or insufficient T cell trafficking to the tumor sites.Citation86,Citation87
Due to the low frequency of tumor-reactive T cells present in the peripheral blood of cancer patients, enrichment of antigen-specific T cells is essential in ACT manufacturing. Our group has demonstrated that RNA electroporation selectively targets gene expression to actively dividing T cells and, thus, transfection of reporter genes such as GFP can be used to isolate and expand antigen-specific T cells responding to antigens of interest for use in ACT.Citation55
Based on recent data reporting the direct effects of toll-like receptor (TLR) ligands on effector T cell function, Citation88 Pato et al. electroporated tumor-infiltrating lymphocytes from melanoma patients with a constitutively activated TLR-4 encoding mRNA, which prolonged the expression of activation markers, induced the secretion of cytokines and chemokines, and increased effector T cell function.Citation51 In another study, Kunni et al. replaced the integral membrane adapter molecule linker for activation of T cells (LAT), known to play a central role in the activation of primary human CD4+ and CD8+ T cells, with ubiquitylation-resistant LAT (LAT 2KR). This substitution resulted in enhanced T cell signaling, proliferation, and Th1 cytokine secretion. By transfecting anti-CD19-ζ CAR T cells or anti-NY-ESO-1 TCR T cells with LAT 2KR mRNA, the authors demonstrated increased CAR and TCR T cell-mediated antitumor efficacy in vitro.Citation89 A recent major obstacle in the efficiency of engineered T cells against solid tumors is the upregulation of T-cell inhibitory receptors (e.g., PD‐1 and CTLA‐4), resulting in limited antitumor response. By simultaneously electroporating T cells with siRNA and mRNA encoding for PD-1/CTLA-4 and CAR specific for chondroitin sulfate proteoglycan 4, respectively, Simon et al. demonstrated superior in vitro CAR T cell functions against melanoma cells compared with CAR T cell alone.Citation22 Overall, these studies demonstrate that various RNA-mediated modifications can be employed to enhance T cell effector functions.
Improvement of T cell migration and persistence
The ex vivo expansion of antigen-specific T cells for ACT often results in highly activated or exhausted T cell phenotypes, Citation90,Citation91 leading to reduced T cell trafficking and persistence at the tumor siteCitation92 and limiting clinical efficacy. Our group has previously demonstrated that the electroporation of antigen-specific T cells with RNA encoding for chemokine receptor CXCR2 leads to the efficient migration of these modified T cells toward glioma-secreted CXCR2-specific ligands in vitro and in vivo, Citation55 showing the possibility of using RNA modifications to enhance the chemotactic functions of T cells.
Despite the genetic stability of engineered T cells generated using viral vectors, these T cells can become exhausted by the time sufficient number of transfectants are obtained due to the long-term selection process involved in the utilization of those vectors. This process can result in T cell telomere shortening, inefficient proliferation, and short-term persistence in vivo.Citation93 Consequently, transient RNA T cell modifications have been used to complement viral vector-mediated CAR and TCR modifications in order to enhance the persistence of transferred T cells.Citation36,Citation38,Citation53 By transfecting anti-CD19-expressing CAR T cells with RNA encoding for telomerase reverse transcriptase (TERT), a major protein directly associated with cellular senescence, Bai et al. have demonstrated that TERT CD19 CAR T cells display transiently enhanced telomerase activity, increased telomere length, and decreased senescence. Notably, these cells displayed long-term antitumor immune responses in vivo.Citation38
The destruction of transferred T cells by lymphodepleting agents, allogeneic/“off-the-shelf” manufacturing, or immune rejection by the host have created major concerns about the utilization of ACT in cancer patients.Citation94 By deleting T cells’ αβ TCR [which mediates graft-versus-host disease (GvHD)] using transcription activator-like effector nuclease (TALEN) and CD52, a protein targeted by the monoclonal antibody alemtuzumab, Poirot et al. hypothesized that functional allogeneic antigen-specific T cells could be manufactured, while avoiding GvHD responses and rendering T cells resistant to destruction by alemtuzumab.Citation36 Upon exposure to alemtuzumab, CD52 and TALEN RNA-co-transfected CD19 CAR T cells displayed efficient CD19+ tumor cell killing compared to unmodified CD19 CAR T cells.Citation36 Together, these studies demonstrate the utility of RNA transfection as a simple and efficient method to enhance the persistence and migration of tumor-reactive T cells.
Improvement of RNA stability and duration
Upon tumor antigen recognition, activated T cells undergo homeostatic proliferation as well as RNA metabolism and degradation; notably, RNA expression declines more rapidly in vivo than in vitro, potentially hindering the long-term therapeutic effects of RNA-modified T cells. While transient transgene expression may be desirable in certain cases, it could also be a limitation for prolonged therapeutic delivery. Therefore, researchers have been exploring methods to improve stability and duration of transgene expression following RNA electroporation in T cells.
The in vitro synthesis and transfection efficiency of mRNA have been shown to depend on its structure (e.g., the capping nucleotide and poly (A) tail).Citation42 To improve the stability of mRNAs encoding CARs, Zhao et al. have tested the insertion of an α-globin 3ʹ untranslated region (prolonged (150A) poly (A) sequence) and an anti-reverse cap analogue or cap1 structure.Citation27 While these modifications led to increased transgene expression efficiency, no effects on transgene expression length were observed.Citation27 Furthermore, to reduce potential harmful “off-target” translation products and avoid immune rejection of engineered T cells, IVT RNA has been codon optimized by removing the open reading frame (ORF).Citation25 For instance, compared to parental FRα-specific CAR RNA T cells, optimized CAR RNA T cells demonstrated similar immunoreactivity, cytolytic potential, and antitumor function.Citation25 Therefore, while the limited duration of transgene expression following mRNA transfection may allow for safety when using RNA-modified T cells alone or in combination with current viral vector-based T cell engineering, there may be a need to optimize the RNA construct.
Conclusion and future directions
Ex vivo T cell expansion has permitted the manipulation of T cells prior to ACT for cancer, and extensive investigations have been performed on RNA-mediated T cell modifications to improve T-cell antigen specificity, trafficking, and functions. More recently, RNA-electroporated T cells have proven to be an effective tool for immunomodulatory agent delivery for cancer therapy.Citation52,Citation56 Compared to viral vectors, clinical-grade synthetic mRNA can easily be generated in large quantities and in a cell-free process, allowing for minimal costs and regulations. Several RNA-modified T cell therapy approaches are currently being tested in clinical trials for patients with advanced malignancies. In addition, methods to improve RNA stability, purity, and duration have been investigated in order to enhance the therapeutic efficacy of those modified T cells. An optimal treatment regimen remains to be determined, including dose and number of T cell injections, T cell population to transfect, and type of RNA used for T cell modifications in order to achieve optimal therapeutic effects.
Although ACT using RNA-modified T cells has proven to be a promising therapeutic tool for hematologic malignancies, developments are still needed for its application in the treatment of solid tumors, mostly due to the lack of target tumor antigens that are not expressed by normal tissues, tumor localization, and immunosuppressive microenvironment. While unprecedented durable response rates have been observed with ACT, most patients do not benefit from the treatment (primary resistance, e.g., due to the absence of tumor antigens, such as in pancreatic cancer), and some responders relapse after a period of response (acquired resistance, e.g., due to the development of mechanisms to avoid antigen presentation on surface-restricted MHC). To overcome these obstacles, combinatorial approaches with other types of treatment, such as chemo-, radio-, and targeted therapies, have been explored.Citation95–Citation102 In addition, suppressive cytokines/agents and cell populations such as myeloid-derived suppressor cells and regulatory T cells present in the tumor microenvironment can induce immunosuppression, thereby blocking T cell function.Citation103,Citation104 Therefore, combining ACT therapy with other strategies that inhibit negative regulators such as PD-1/PD-L1, or reverse regulatory T cell/myeloid-derived suppressor cell-mediated immunosuppression, may induce potentially profound immune antitumor responses.Citation105–Citation110 Furthermore, the combination of T cell ACT with other forms of immune cell therapies has also yielded very promising results in preclinical studies.Citation111,Citation112 Finally, for those combinatorial treatments to be implemented on a large scale for various cancer patient populations, issues pertaining to scale‐up, automation, commercialization, intellectual property, costs/insurance reimbursements, and regulatory hurdles unique to cell therapy will need to be addressed.Citation1,Citation113,Citation114
Disclosure of potential conflicts of interest
DAM holds ownership interest (including patents) in iOncologi, Inc., and DAM has patented immunotherapy-related technology that has been licensed by Annias Immunotherapeutics, Inc.; Celldex Therapeutics, Inc.; and Immunomic Therapeutics, Inc. FPG and LBH declare no conflicts of interest.
Acknowledgments
We would like to thank Laura Dickinson for her diligent proofreading of this paper.
Additional information
Funding
References
- Ruella M, Kalos M. Adoptive immunotherapy for cancer. Immunol Rev. 2014;257:14–13.
- Leavy O. Therapeutic antibodies: past, present and future. Nat Rev Immunol. 2010;10(5):297. doi:10.1038/nri2763.
- Darvin P, Toor SM, Nair VS, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers.
- Weiner LM, Murray JC, Shuptrine CW. Antibody-based immunotherapy of cancer. Cell. 2012;148(6):1081–1084. doi:10.1016/j.cell.2012.02.034.
- Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7(9):987–989. doi:10.1038/nm0901-987.
- Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. doi:10.1056/NEJMoa1305133.
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi:10.1056/NEJMoa1200690.
- Sandin LC, Eriksson F, Ellmark P, Loskog AS, Tötterman TH, Mangsbo SM. Local CTLA4 blockade effectively restrains experimental pancreatic adenocarcinoma growth in vivo. Oncoimmunology. 2014;3(1):e27614. doi:10.4161/onci.27614.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736. doi:10.1126/science.271.5256.1734.
- Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. doi:10.1056/NEJMoa1504627.
- Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23(10):2346–2357. doi:10.1200/JCO.2005.00.240.
- Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3(9):666–675. doi:10.1038/nrc1167.
- Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850–854. doi:10.1126/science.1076514.
- Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319(25):1676–1680. doi:10.1056/NEJM198812223192527.
- Aoki Y, Takakuwa K, Kodama S, Tanaka K, Takahashi M, Tokunaga A, Takahashi T. Use of adoptive transfer of tumor-infiltrating lymphocytes alone or in combination with cisplatin-containing chemotherapy in patients with epithelial ovarian cancer. Cancer Res. 1991;51:1934–1939.
- Freedman RS, Edwards CL, Kavanagh JJ, Kudelka AP, Katz RL, Carrasco CH, Atkinson EN, Scott W, Tomasovic B, Templin S. Intraperitoneal adoptive immunotherapy of ovarian carcinoma with tumor-infiltrating lymphocytes and low-dose recombinant interleukin-2: a pilot trial. J Immunother Emphasis Tumor Immunol. 1994;16(3):198–210. doi:10.1097/00002371-199410000-00004.
- Fujita K, Ikarashi H, Takakuwa K, Kodama S, Tokunaga A, Takahashi T, Tanaka K. Prolonged disease-free period in patients with advanced epithelial ovarian cancer after adoptive transfer of tumor-infiltrating lymphocytes. Clin Cancer Res. 1995;1:501–507.
- Fabbri M, Ridolfi R, Maltoni R, Ridolfi L, Riccobon A, Flamini E, De Paola F, Verdecchia GM, Amadori D. Tumor infiltrating lymphocytes and continuous infusion interleukin-2 after metastasectomy in 61 patients with melanoma, colorectal and renal carcinoma. Tumori Journal. 2000;86(1):46–52. doi:10.1177/030089160008600109.
- Topalian SL, Solomon D, Avis FP, Chang AE, Freerksen DL, Linehan WM, Lotze MT, Robertson CN, Seipp CA, Simon P. Immunotherapy of patients with advanced cancer using tumor-infiltrating lymphocytes and recombinant interleukin-2: a pilot study. J Clin Oncol. 1988;6(5):839–853. doi:10.1200/JCO.1988.6.5.839.
- Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce antitumor activity in solid malignancies. Cancer Immunol Res. 2014;2(2):112–120. doi:10.1158/2326-6066.CIR-13-0170.
- Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJD, Scholler J, Song D, Porter DL, Carroll M, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29(8):1637–1647. doi:10.1038/leu.2015.52.
- Simon B, Harrer DC, Schuler-Thurner B, Schaft N, Schuler G, Dörrie J, Uslu U. The siRNA-mediated downregulation of PD-1 alone or simultaneously with CTLA-4 shows enhanced in vitro CAR-T-cell functionality for further clinical development towards the potential use in immunotherapy of melanoma. Exp Dermatol. 2018;27(7):769–778. doi:10.1111/exd.13678.
- Uslu U, Schuler G, Dörrie J, Schaft N. Combining a chimeric antigen receptor and a conventional T-cell receptor to generate T cells expressing two additional receptors (TETARs) for a multi-hit immunotherapy of melanoma. Exp Dermatol. 2016;25(11):872–879. doi:10.1111/exd.13095.
- Caruso HG, Torikai H, Zhang L, Maiti S, Dai J, Do K-A, Singh H, Huls H, Lee DA, Champlin RE. Redirecting T-Cell specificity to EGFR using mRNA to self-limit expression of chimeric antigen receptor. J Immunother. 2016;39(5):205–217. doi:10.1097/CJI.0000000000000126.
- Schutsky K, Song D-G, Lynn R, Smith JB, Poussin M, Figini M, Zhao Y, Powell DJ. Rigorous optimization and validation of potent RNA CAR T cell therapy for the treatment of common epithelial cancers expressing folate receptor. Oncotarget. 2015;6(30):28911–28928. doi:10.18632/oncotarget.5029.
- Hofflin S, Prommersberger S, Uslu U, Schuler G, Schmidt CW, Lennerz V, Dörrie J, Schaft N. Generation of CD8+ T cells expressing two additional T-cell receptors (TETARs) for personalised melanoma therapy. Cancer Biol Ther. 2015;16(9):1323–1331. doi:10.1080/15384047.2015.1070981.
- Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, Chew A, Carroll RG, Scholler J, Levine BL. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70(22):9053–9061. doi:10.1158/0008-5472.CAN-10-2880.
- Parkhurst MR, Joo J, Riley JP, Yu Z, Li Y, Robbins PF, Rosenberg SA. Characterization of genetically modified T-cell receptors that recognize the CEA:691-699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. Clin Cancer Res. 2009;15(1):169–180. doi:10.1158/1078-0432.CCR-08-1638.
- Birkholz K, Hombach A, Krug C, Reuter S, Kershaw M, Kämpgen E, Schuler G, Abken H, Schaft N, Dörrie J. Transfer of mRNA encoding recombinant immunoreceptors reprograms CD4+ and CD8+ T cells for use in the adoptive immunotherapy of cancer. Gene Therapy. 2009;16(5):596–604. doi:10.1038/gt.2008.189.
- Yoon SH, Lee JM, Cho HI, Kim EK, Kim HS, Park MY, Kim TG. Adoptive immunotherapy using human peripheral blood lymphocytes transferred with RNA encoding Her-2/neu-specific chimeric immune receptor in ovarian cancer xenograft model. Cancer Gene Ther. 2009;16(6):489–497. doi:10.1038/cgt.2008.98.
- Schaft N. A new way to generate cytolytic tumor-specific T cells: electroporation of RNA coding for a T cell receptor into T lymphocytes. Cancer Immunol Immunother. 2006;55(9):1132–1141. doi:10.1007/s00262-005-0098-2.
- Zhao Y, Zheng Z, Robbins PF, Khong HT, Rosenberg SA, Morgan RA. Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J Immunol. 2005;174(7):4415–4423. doi:10.4049/jimmunol.174.7.4415.
- Campillo-Davo D, Fujiki F, Van den Bergh JMJ, De Reu H, Smits ELJM, Goossens H, Sugiyama H, Lion E, Berneman ZN, Van Tendeloo V. Efficient and non-genotoxic RNA-based engineering of human T cells using tumor-specific T cell receptors with minimal TCR mispairing. Front Immunol. 2018;9:2503. doi:10.3389/fimmu.2018.02503.
- Liu X, Barrett DM, Jiang S, Fang C, Kalos M, Grupp SA, June CH, Zhao Y. Improved anti-leukemia activities of adoptively transferred T cells expressing bispecific T-cell engager in mice. Blood Cancer Journal. 2016;6(6):e430. doi:10.1038/bcj.2016.38.
- Panjwani MK, Smith JB, Schutsky K, Gnanandarajah J, O’Connor CM, Powell DJ, Mason NJ. Feasibility and safety of RNA-transfected CD20-specific chimeric antigen receptor T cells in dogs with spontaneous B cell lymphoma. Mol Ther. 2016;24(9):1602–1614. doi:10.1038/mt.2016.146.
- Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, Potrel P, Bas C, Lemaire L, Galetto R. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 2015;75(18):3853–3864. doi:10.1158/0008-5472.CAN-14-3321.
- Shimizu K, Shinga J, Yamasaki S, Kawamura M, Dörrie J, Schaft N, Sato Y, Iyoda T, Fujii S. Transfer of mRNA encoding invariant NKT cell receptors imparts glycolipid specific responses to T cells and gammadeltaT cells. PLoS One. 2015;10(6):e0131477. doi:10.1371/journal.pone.0131477.
- Bai Y, Kan S, Zhou S, Wang Y, Xu J, Cooke JP, Wen J, Deng H. Enhancement of the in vivo persistence and antitumor efficacy of CD19 chimeric antigen receptor T cells through the delivery of modified TERT mRNA. Cell Discov. 2015;1:15040. doi:10.1038/celldisc.2015.40.
- Kunii N, Zhao Y, Jiang S, Liu X, Scholler J, Balagopalan L, Samelson LE, Milone MC, June CH. Enhanced function of redirected human T cells expressing linker for activation of T cells that is resistant to ubiquitylation. Hum Gene Ther. 2013;24(1):27–37. doi:10.1089/hum.2012.130.
- Barrett DM, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther. 2013;24(8):717–727. doi:10.1089/hum.2013.075.
- Rabinovich PM, Komarovskaya ME, Wrzesinski SH, Alderman JL, Budak-Alpdogan T, Karpikov A, Guo H, Flavell RA, Cheung N-K, Weissman SM. Chimeric receptor mRNA transfection as a tool to generate antineoplastic lymphocytes. Hum Gene Ther. 2009;20(1):51–61. doi:10.1089/hum.2008.068.
- Rabinovich PM, Komarovskaya ME, Ye Z-J, Imai C, Campana D, Bahceci E, Weissman SM. Synthetic messenger RNA as a tool for gene therapy. Hum Gene Ther. 2006;17(10):1027–1035. doi:10.1089/hum.2006.17.1027.
- Nienhuis AW, Dunbar CE, Sorrentino BP. Genotoxicity of retroviral integration in hematopoietic cells. Mol Ther. 2006;13(6):1031–1049. doi:10.1016/j.ymthe.2006.03.001.
- Baum C, Kustikova O, Modlich U, Li Z, Fehse B. Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum Gene Ther. 2006;17(3):253–263. doi:10.1089/hum.2006.17.253.
- Yi Y, Hahm SH, Lee KH. Retroviral gene therapy: safety issues and possible solutions. Curr Gene Ther. 2005;5(1):25–35. doi:10.2174/1566523052997514.
- Qin D-Y, Huang Y, Li D, Wang Y-S, Wang W, Wei Y-Q. Paralleled comparison of vectors for the generation of CAR-T cells. Anticancer Drugs. 2016;27(8):711–722. doi:10.1097/CAD.0000000000000387.
- Riet T, Holzinger A, Dörrie J, Schaft N, Schuler G, Abken H. Nonviral RNA transfection to transiently modify T cells with chimeric antigen receptors for adoptive therapy. Methods Mol Biol. 2013;969:187–201.
- Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. doi:10.1126/science.1129003.
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi:10.1056/NEJMoa1103849.
- Ang WX, Li Z, Chi Z, Du S-H, Chen C, Tay JCK, Toh HC, Connolly JE, Xu XH, Wang S. Intraperitoneal immunotherapy with T cells stably and transiently expressing anti-EpCAM CAR in xenograft models of peritoneal carcinomatosis. Oncotarget. 2017;8(8):13545–13559. doi:10.18632/oncotarget.14592.
- Pato A, Eisenberg G, Machlenkin A, Margalit A, Cafri G, Frankenburg S, Merims S, Peretz T, Lotem M, Gross G, et al. Messenger RNA encoding constitutively active Toll-like receptor 4 enhances effector functions of human T cells. Clin Exp Immunol. 2015;182(2):220–229. doi:10.1111/cei.12688.
- Pohl-Guimarães F, Yang C, Dyson KA, Wildes TJ, Drake J, Huang J, Flores C, Sayour EJ, Mitchell DA. RNA-modified T cells mediate effective delivery of immunomodulatory cytokines to brain tumors. 2019;27(4):837–849.
- Almasbak H, Rian E, Hoel HJ, Pulè M, Wälchli S, Kvalheim G, Gaudernack G, Rasmussen A-M. Transiently redirected T cells for adoptive transfer. Cytotherapy. 2011;13(5):629–640. doi:10.3109/14653249.2010.542461.
- Barrett DM, Zhao Y, Liu X, Jiang S, Carpenito C, Kalos M, Carroll RG, June CH, Grupp SA. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Ther. 2011;22(12):1575–1586. doi:10.1089/hum.2011.070.
- Mitchell DA, Karikari I, Cui X, Xie W, Schmittling R, Sampson JH. Selective modification of antigen-specific T cells by RNA electroporation. Hum Gene Ther. 2008;19(5):511–521. doi:10.1089/hum.2007.115.
- Eggers R, Philippi A, Altmeyer MO, Breinig F, Schmitt MJ. Primary T cells for mRNA-mediated immunotoxin delivery. Gene Ther. 2018;25(1):47–53. doi:10.1038/gt.2017.87.
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. doi:10.1016/j.immuni.2013.07.012.
- Johnson BA 3rd, Yarchoan M, Lee V, Laheru DA, Jaffee EM. Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin Cancer Res. 2017;23(7):1656–1669. doi:10.1158/1078-0432.CCR-16-2318.
- Chen S, Lee L-F, Fisher TS, Jessen B, Elliott M, Evering W, Logronio K, Tu GH, Tsaparikos K, Li X. Combination of 4-1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol Res. 2015;3(2):149–160. doi:10.1158/2326-6066.CIR-14-0118.
- Cho D, Kim T-G, Lee W, Hwang Y-I, Cho H-I, Han H, Kim D, Kwon O, Park H, Houh D, et al. Interleukin-18 and the costimulatory molecule B7-1 have a synergistic anti-tumor effect on murine melanoma; implication of combined immunotherapy for poorly immunogenic malignancy. J Invest Dermatol. 2000;114(5):928–934. doi:10.1038/sj.jid.5600685.
- Naguma E, Kawashima K, Nagase F, Nakashima I, Ohno R, Yamada K. Adoptive immunotherapy against a local or widespread tumor of a highly malignant and poorly immunogenic mouse leukemia L1210. Nihon Ketsueki Gakkai Zasshi. 1986;49:1212–1221.
- Reiser J, Banerjee A. Effector, memory, and dysfunctional CD8(+) T cell fates in the antitumor immune response. J Immunol Res. 2016;2016:8941260. doi:10.1155/2016/8941260.
- Theobald M, Biggs J, Hernández J, Lustgarten J, Labadie C, Sherman LA. Tolerance to p53 by A2.1-restricted cytotoxic T lymphocytes. J Exp Med. 1997;185(5):833–841. doi:10.1084/jem.185.5.833.
- Lippitz BE. Cytokine patterns in patients with cancer: a systematic review. Lancet Oncol. 2013;14(6):e218–28. doi:10.1016/S1470-2045(12)70582-X.
- Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–924. doi:10.1200/JCO.2010.32.2537.
- Sun S, Hao H, Yang G, Zhang Y, Fu Y. Immunotherapy with CAR-modified T cells: toxicities and overcoming strategies. J Immunol Res. 2018;2018:2386187. doi:10.1155/2018/2386187.
- Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. doi:10.1126/scitranslmed.3008226.
- Heiser A, Maurice MA, Yancey DR, Wu NZ, Dahm P, Pruitt SK, Boczkowski D, Nair SK, Ballo MS, Gilboa E, et al. Induction of polyclonal prostate cancer-specific CTL using dendritic cells transfected with amplified tumor RNA. J Immunol. 2001;166(5):2953–2960. doi:10.4049/jimmunol.166.5.2953.
- Mitchell DA, Nair SK. RNA-transfected dendritic cells in cancer immunotherapy. J Clin Invest. 2000;106(9):1065–1069. doi:10.1172/JCI11405.
- Nair SK, Heiser A, Boczkowski D, Majumdar A, Naoe M, Lebkowski JS, Vieweg J, Gilboa E. Induction of cytotoxic T cell responses and tumor immunity against unrelated tumors using telomerase reverse transcriptase RNA transfected dendritic cells. Nat Med. 2000;6(9):1011–1017. doi:10.1038/79519.
- Hurwitz AA, Cuss SM, Stagliano KE, Zhu Z. T cell avidity and tumor immunity: problems and solutions. Cancer Microenviron. 2014;7(1–2):1–9. doi:10.1007/s12307-013-0143-1.
- van der Veken LT, Hagedoorn RS, van Loenen MM, Willemze R, Falkenburg JHF, Heemskerk MHM. Alphabeta T-cell receptor engineered gammadelta T cells mediate effective antileukemic reactivity. Cancer Res. 2006;66(6):3331–3337. doi:10.1158/0008-5472.CAN-05-4190.
- Reeves E, James E. Antigen processing and immune regulation in the response to tumours. Immunology. 2017;150(1):16–24. doi:10.1111/imm.12675.
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851. doi:10.1038/mt.2010.24.
- Singh N, Liu X, Hulitt J, Jiang S, June CH, Grupp SA, Barrett DM, Zhao Y. Nature of tumor control by permanently and transiently modified GD2 chimeric antigen receptor T cells in xenograft models of neuroblastoma. Cancer Immunol Res. 2014;2(11):1059–1070. doi:10.1158/2326-6066.CIR-14-0051.
- Singh N, Barrett DM, Grupp SA. Roadblocks to success for RNA CARs in solid tumors. Oncoimmunology. 2014;3(12):e962974. doi:10.4161/21624011.2014.962974.
- Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21(4):687–692. doi:10.1158/1078-0432.CCR-14-1860.
- Sadun RE, Sachsman SM, Chen X, Christenson KW, Morris WZ, Hu P, Epstein AL. Immune signatures of murine and human cancers reveal unique mechanisms of tumor escape and new targets for cancer immunotherapy. Clin Cancer Res. 2007;13(13):4016–4025. doi:10.1158/1078-0432.CCR-07-0016.
- Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290–296. doi:10.1038/icb.2014.93.
- Litzow MR. Monoclonal antibody-based therapies in the treatment of acute lymphoblastic leukemia. Am Soc Clin Oncol Educ Book. 2013;294–299. doi:10.14694/EdBook_AM.2013.33.294.
- Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, Nichols KE, Suppa EK, Kalos M, Berg RA. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121(26):5154–5157. doi:10.1182/blood-2013-02-485623.
- Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8(6):467–477. doi:10.1038/nri2326.
- Wintterle S, Schreiner B, Mitsdoerffer M, Schneider D, Chen L, Meyermann R, Weller M, Wiendl H. Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res. 2003;63:7462–7467.
- Huettner C, Czub S, Kerkau S, Roggendorf W, Tonn JC. Interleukin 10 is expressed in human gliomas in vivo and increases glioma cell proliferation and motility in vitro. Anticancer Res. 1997;17(5A:3217–3224.
- Zuber P, Kuppner MC, De Tribolet N. Transforming growth factor-beta 2 down-regulates HLA-DR antigen expression on human malignant glioma cells. Eur J Immunol. 1988;18(10):1623–1626. doi:10.1002/eji.1830181023.
- Peng W, Liu C, Xu C, Lou Y, Chen J, Yang Y, Yagita H, Overwijk WW, Lizee G, Radvanyi L. PD-1 blockade enhances T-cell migration to tumors by elevating IFN-gamma inducible chemokines. Cancer Res. 2012;72(20):5209–5218. doi:10.1158/0008-5472.CAN-12-1187.
- Franciszkiewicz K, Boissonnas A, Boutet M, Combadiere C, Mami-Chouaib F. Role of chemokines and chemokine receptors in shaping the effector phase of the antitumor immune response. Cancer Res. 2012;72(24):6325–6332. doi:10.1158/0008-5472.CAN-12-2027.
- Reynolds JM, Dong C. Toll-like receptor regulation of effector T lymphocyte function. Trends Immunol. 2013;34(10):511–519. doi:10.1016/j.it.2013.06.003.
- Balagopalan L, Ashwell BA, Bernot KM, Akpan IO, Quasba N, Barr VA, Samelson LE. Enhanced T-cell signaling in cells bearing linker for activation of T-cell (LAT) molecules resistant to ubiquitylation. Proc Natl Acad Sci U S A. 2011;108(7):2885–2890. doi:10.1073/pnas.1007098108.
- Dudley ME, Wunderlich JR, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry RM, Marincola FM, Leitman SF, Seipp CA. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother. 2002;25(3):243–251. doi:10.1097/00002371-200205000-00007.
- Dudley ME, Wunderlich J, Nishimura MI, Yu D, Yang JC, Topalian SL, Schwartzentruber DJ, Hwu P, Marincola FM, Sherry R, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother. 2001;24(4):363–373. doi:10.1097/00002371-200107000-00012.
- Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A. 2002;99(25):16168–16173. doi:10.1073/pnas.242600099.
- Zhou J, Shen X, Huang J, Hodes RJ, Rosenberg SA, Robbins PF. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J Immunol. 2005;175(10):7046–7052. doi:10.4049/jimmunol.175.10.7046.
- Morris EC, Rebello P, Thomson KJ, Peggs KS, Kyriakou C, Goldstone AH, Mackinnon S, Hale G. Pharmacokinetics of alemtuzumab used for in vivo and in vitro T-cell depletion in allogeneic transplantations: relevance for early adoptive immunotherapy and infectious complications. Blood. 2003;102(1):404–406. doi:10.1182/blood-2002-09-2687.
- Karlsson H, Lindqvist AC, Fransson M, Paul-Wetterberg G, Nilsson B, Essand M, Nilsson K, Frisk P, Jernberg-Wiklund H, Loskog SIA. Combining CAR T cells and the Bcl-2 family apoptosis inhibitor ABT-737 for treating B-cell malignancy. Cancer Gene Ther. 2013;20(7):386–393. doi:10.1038/cgt.2013.35.
- Sanchez C, Chan R, Bajgain P, Rambally S, Palapattu G, Mims M, Rooney CM, Leen AM, Brenner MK, Vera JF, et al. Combining T-cell immunotherapy and anti-androgen therapy for prostate cancer. Prostate Cancer Prostatic Dis. 2013;16(2):123–31 S1. doi:10.1038/pcan.2012.49.
- Patel KK, Olivares S, Singh H, Hurton LV, Huls MH, Qazilbash MH, Kebriaei P, Champlin RE, Cooper LJN. Combination Immunotherapy with NY-ESO-1-Specific CAR(+) T Cells with T-Cell Vaccine Improves Anti-Myeloma Effect.
- Muranski P, Boni A, Wrzesinski C, Citrin DE, Rosenberg SA, Childs R, Restifo NP. Increased intensity lymphodepletion and adoptive immunotherapy - how far can we go? Nat Clin Pract Oncol. 2006;3(12):668–681. doi:10.1038/ncponc0666.
- Rosenberg SA, Dudley ME. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol. 2009;21(2):233–240. doi:10.1016/j.coi.2009.03.002.
- Vierboom MP, Bos GMJ, Ooms M, Offringa R, Melief CJM. Cyclophosphamide enhances anti-tumor effect of wild-type p53-specific CTL. Int J Cancer. 2000;87(2):253–260. doi:10.1002/1097-0215(20000715)87:2<253::AID-IJC17>3.0.CO;2-A.
- Parente-Pereira AC, Whilding LM, Brewig N, van der Stegen SJC, Davies DM, Wilkie S, van Schalkwyk MCI, Ghaem-Maghami S, Maher J. Synergistic chemoimmunotherapy of epithelial ovarian cancer using ErbB-retargeted T cells combined with carboplatin. The Journal of Immunology. 2013;191(5):2437–2445. doi:10.4049/jimmunol.1301119.
- Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-based CAR T cells and radiotherapy exert synergistic efficacy in glioblastoma. Cancer Res. 2018;78(4):1031–1043. doi:10.1158/0008-5472.CAN-17-1788.
- Wang HY, Wang RF. Regulatory T cells and cancer. Curr Opin Immunol. 2007;19(2):217–223. doi:10.1016/j.coi.2007.02.004.
- Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3(+) regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10(7):490–500. doi:10.1038/nri2785.
- Locke FL, Westin JR, Miklos DB, Herrara AF, Jacobson CA, Lee J, Rossi JM, Bot A, Xue A, Navale L, et al. Phase 1 results from ZUMA-6: axicabtagene Ciloleucel (axi-cel; KTE-C19) in combination with atezolizumab for the treatment of patients with refractory diffuse large B cell lymphoma (DLBCL).
- Moon EK, Wang L-C, Dolfi DV, Wilson CB, Ranganathan R, Sun J, Kapoor V, Scholler J, Pure E, Milone MC. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clinical Cancer Research. 2014;20(16):4262–4273. doi:10.1158/1078-0432.CCR-13-2627.
- Yoon DH, Osborn MJ, Tolar J, Kim CJ. Incorporation of immune checkpoint blockade into chimeric antigen receptor T cells (CAR-Ts): combination or Built-In CAR-T.
- Oelkrug C, Ramage JM. Enhancement of T cell recruitment and infiltration into tumours. Clin Exp Immunol. 2014;178(1):1–8. doi:10.1111/cei.12382.
- Esposito A, Criscitiello C, Curigliano G. Immune checkpoint inhibitors with radiotherapy and locoregional treatment: synergism and potential clinical implications. Curr Opin Oncol. 2015;27(6):445–451. doi:10.1097/CCO.0000000000000225.
- Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, June CH, Schuster SJ. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood. 2017;129(8):1039–1041. doi:10.1182/blood-2016-09-738245.
- Flores CT, Wildes TJ, Drake JA, Moore GL, Dean BD, Abraham RS, Mitchell DA. Lin(-)CCR2(+) hematopoietic stem and progenitor cells overcome resistance to PD-1 blockade1. Nat Commun. 2018;9. doi:10.1038/s41467-018-06182-5.
- Wildes TJ, Grippin A, Dyson KA, Wummer BM, Damiani DJ, Abraham RS, Flores CT, Mitchell DA. Cross-talk between T cells and hematopoietic stem cells during adoptive cellular therapy for malignant glioma. Clinical Cancer Research. 2018;24(16):3955–3966. doi:10.1158/1078-0432.CCR-17-3061.
- Kohl U, Arsenieva S, Holzinger A, Abken H. CAR T cells in trials: recent achievements and challenges that remain in the production of modified T cells for clinical applications. Hum Gene Ther. 2018;29(5):559–568. doi:10.1089/hum.2017.254.
- Srivastava S, Riddell SR. Chimeric antigen receptor T cell therapy: challenges to bench-to-bedside efficacy. The Journal of Immunology. 2018;200(2):459–468. doi:10.4049/jimmunol.1701155.