
ABSTRACT
The tumoricidal efficiency of human CAR-T cells is generally evaluated using immune-deficient mouse models; however, due to their immune-incompetency and the species-specific reactivity of a target antigen, these models are problematic to imitate CAR-T-induced adverse effects in the clinic. Epithelial cell adhesion molecule (EpCAM) is a tumor-associated antigen overtly presented on the cell surface of various carcinomas, making it an attractive target for CAR-T therapy. Here, we developed an anti-mouse EpCAM CAR to evaluate its safety and efficacy in immunocompetent mouse models. As previously reported for their human equivalents, murine EpCAM CAR-T cells exhibit promising anti-tumor efficacy in vitro and in vivo. However, after CAR-T infusion, various dose-depended toxicities including body weight loss, cytokine-release syndrome (CRS), and death were observed in both tumor-bearing and tumor-free mice. Pathological examination revealed unexpected and severe pulmonary immunopathology due to basal EpCAM expression in normal lung. While our study validates EpCAM CAR-T’s potent anti-tumor efficacy, it also reveals that EpCAM CAR-T cells used for the treatment of solid tumors may cause lethal toxicity and should, therefore, be evaluated in patients with caution.
Introduction
Genetically engineered human T cells expressing a chimeric antigen receptor (CAR) have been revolutionary for cancer treatment. In particular, CD19 CAR-T cells have shown impressive therapeutic responses (70–94% CR) against acute and chronic leukemia and B lineage non-Hodgkin lymphoma (NHL),1-Citation5 such that two CD19 CAR-T products are now approved by the US Food and Drug Administration for the treatment of select hematologic malignancies.Citation6,Citation7 This success of CD19 CAR-T has inspired researchers to explore the application of other antigen-specific CAR-T cells as new therapies, especially for solid tumors.
Two main issues must be considered before initiating a CAR-T clinical trial. One is anti-tumor efficacy, which is the primary purpose of treatment. In the preclinical stage, the anti-tumor effect of human CAR-T cells can be assessed using in vitro assays or in immune-deficient mouse models such as NSG mice bearing human-derived tumors; this can predict the CAR-T’s therapeutic efficacy to some extent. The other, and more preeminent one, is the toxicity caused by CAR-T therapy, which is thought to result from the systemic activation of CAR-T cells following engagement of target antigen expressed on tumor cells or normal tissues.Citation8–Citation10 Those activated CAR-T cells can further initiate a series of immune reactions, such as activating monocytes and macrophages,Citation11,Citation12 resulting in fatal cytokine-release syndrome (CRS), or directly attack antigen-expressing normal tissues (on-target/off-tumor side effect). Currently available preclinical xenograft mouse models can be used to evaluate CAR-T cells’ anti-tumor efficacy, but are poorly predictive of their potential toxicity due to these animals’ incomplete immune system.Citation13,Citation14 Monocytes and macrophages are necessary for CRS induction, while the development and maturation of these cells are defective in NOD mice.Citation11 The IL2Rγ mutation present in NSG mice also impairs IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 cytokine signaling,Citation15–Citation17 which negatively impacts T cell differentiation, proliferation, and cell growth.
Epithelial cell adhesion molecule (EpCAM) is a transmembrane glycoprotein which is mainly expressed on certain luminal epithelial cells in an ordered and oriented manner. Upon malignant transformation, EpCAM becomes overexpressed on some carcinomas cells in an unrestricted pattern,Citation18–Citation20 leading to increased accessibility for EpCAM-specific CAR-T cells and heightened CAR-T activation. Apparently, EpCAM has been proposed as an ideal target for CAR-T therapy and existing data demonstrate that human EpCAM targeted CAR-T cells can eradicate established tumor xenografts without toxicities in mouse models.Citation21–Citation23 Based on these results, several CAR-T clinical trials targeting EpCAM have already been initiated.Citation24–Citation28 However, although the efficacy evaluation of these pioneer studies in mice may indeed reflect their anti-tumor potential in human, the safety assessment might be problematic: first, the induction of CRS relies on the interaction between exogenous CAR-T cells and an intact mouse immune system; and second, the human EpCAM-specific scFv within these CAR constructs does not recognize mouse autologous EpCAM. Previous clinical trials suggest that the latter is a prominent issue for any CAR targeting a tumor-associated antigen: on-target/off-tumor side effects undetectable in xenograft mouse models may induce patient lethality.Citation10,Citation29
To rigorously assess the safety of EpCAM CAR-T therapy, we developed a CAR construct targeting mouse EpCAM and performed safety and efficacy testing in syngeneic immunocompetent mice. Our data show that mouse T cells armed with this EpCAM-specific CAR display in vivo anti-tumor efficacy similar to that reported for human-derived CAR-T cells. However, we also observed severe dose-dependent toxicity after CAR-T infusion. These results indicate that EpCAM CAR-T cell therapy has a high risk of severe toxicity when applied systemically.
Materials and methods
Mice
Thy1.1+, thy1.2+ B6 (C57BL/6 J), and BALB/c mice were purchased from the Jackson Laboratory and maintained at the animal facility of Duke University under specific pathogen-free conditions. Thy1.1+ thy1.2+ B6 mice were generated by mating B6 albino with thy1.1+ B6 mice. All murine studies were performed in accordance with guidelines and protocols approved by the Duke University Animal Care and Use Committee.
Cell lines
293 T, 3T3, and 4T1 cells were purchased from Duke University Cell Culture Facilities. 293 T and 3T3 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich) supplemented with 10% fetal bovine serum (Atlanta Biologicals) and 1% penicillin/streptomycin (Sigma-Aldrich). 4T1 breast cancer cell lines were cultured in high glucose RPMI 1640 (Sigma-Aldrich) supplemented with 10% fetal bovine serum (Atlanta Biologicals), 1% penicillin/streptomycin (Sigma-Aldrich), 1 mM sodium pyruvate (Gbico), and 10 mM HEPES (Sigma-Aldrich). MC38 mouse colon cancer cells were purchased from the Division of Cancer Treatment and Diagnosis (DCTD) Tumor Repository, and cultured in DMEM with 10% FBS (Atlanta Biologicals) and 1% penicillin/streptomycin (Sigma-Aldrich). All cells were cultured in a 5% CO2 humidified 37° C incubator and tested mycoplasma free before use.
Construction of CAR plasmid and virus production
CAR structures were cloned into a pMSCV backbone. Mouse EpCAM-specific CAR scFv is based on the G8.8 antibody that recognizes the mouse EpCAM antigen.Citation30 The signal peptide, hinge, and transmembrane domains are derived from mouse CD8α, followed by mouse CD28, 41BB, and CD3ζ as costimulatory signaling domains. The mouse EGFRvIII-specific CAR is the same as the EpCAM CAR except for the scFv, which is as described.Citation31 CAR plasmids, along with a pCL-Eco helper plasmid (addgene#12371), were transfected into 293 T cells using Lipofectamine 2000 (Invitrogen) to generate retrovirus. Forty-eight hours after transfection, supernatants were collected and filtered using a 0.45 µm filter.
Murine T cell culture and transduction
T cells were obtained from the lymph nodes of 6–10 week-old mice and cultured in RPMI 1640 supplemented with 10% fetal bovine serum (Atlanta Biologicals), 1% penicillin/streptomycin (Sigma-Aldrich), 2 mM L-glutamine (Gbico), 1 mM sodium pyruvate (Gbico), 0.1 mM nonessential amino acids (Gbico), 0.05 mM 2-mercaptoethanol (Sigma-Aldrich), and 50 U/ml mouse IL-2 (Peprotech). Two µg/ml anti-mCD3 and anti-mCD28 antibody were pre-coated on plates overnight at 4°C to stimulate T cells for 36 h. Activated T cells were spin-infected with retrovirus and Polybrene (6 µg/ml, Sigma-Aldrich) at 2500 rpm 32°C for 90 min. Forty-eight hours after transduction, mouse T cells were used for experiments.
Flow cytometry
For surface staining, cells were first stained with live/dead dye (Invitrogen) and incubated with FcR blocker (2.4G2, BD). The indicated surface-staining antibodies TCRb (H57-597), CD4 (RM4-5), CD8 (53–6.7), Thy1.1 (OX-7), and Thy1.2 (30-H12) (all from BioLegend), were added directly at 4°C for 30 min. EpCAM CAR expression was evaluated by recombinant EpCAM protein tagged with human Fc domain (Novoprotein), which was detected using fluorochrome-conjugated mouse anti-human Fc antibody (BioLegend). EGFRvIII CAR expression using evaluated by biotin-labeled recombinant EGFRvIII peptide (Acro Biosystems), which was detected by fluorochrome-conjugated streptavidin (BioLegend).
For intracellular cytokine staining, Golgi-blocking monensin and brefeldin A (eBioscience) were added 4–6 h before antibody staining. T cells were stained with live/dead dye, FcR blocker, and surface-staining antibodies as described above; then, cells were fixed in 2% paraformaldehyde-PBS and permeabilized in permeabilization buffer (Invitrogen) according to the manufacturer’s protocol. Cells were stained with anti-mouse IFN-γ antibody. Flow cytometry acquisition was performed using a BD FACSCantoTM II analyzer, and FlowJo v10 software (Treestar, Inc. Ashland, OR) was used for data analysis.
In vitro CAR-T proliferation assay
CAR-T cells or control T cells were first incubated with 5 µM CFSE in PBS with 0.2% BSA at 37°C for 10 min; then, 1 × 106 effector cells were co-cultured with 1 × 106 4T1 cells in 6-well plates for 72 h. Cells were stained with fluorochrome-conjugated antibodies (TCRb, CD8, CD4; BioLegend) and analyzed by flow cytometry to detect CFSE dilution. CFSE-labeled control T cells without 4T1 stimulation served as a control.
In vitro CAR-T cytolysis assay
For the LDH detection-based method, effector cells (CAR-T or control T cells) were added to 96-well plates, which contained 1 × 104 target cells (4T1 or 3T3) per well, at increasing effector: target (E:T) ratios (1:1, 2:1, 4:1, 8:1) for 12 h co-culture. Culture supernatants were collected and analyzed using a CytoTox 96® Non-Radioactive Cytotoxicity Assay kit (Promega) according to the manufacturer’s protocol. Percent cytotoxicity was calculated based on the OD value of each group according to the formula: (Experimental – Effector Spontaneous – Target Spontaneous)/(Target Maximum – Target Spontaneous) x 100%.
For the flow cytometry-based method, MC38 cells (5 × 104/well) were labeled with CellTrace violet dye (Invitrogen) and co-cultured with CAR-T or control T cells in 48-well plates at increasing E:T ratios (1:1, 2:1, 4:1, 8:1) for 12 h. MC38 cells without T cells served as a control. All cells in each well were collected in separate tubes, along with 5 µl CountBright™ Absolute Counting Beads (Invitrogen), and stained with live/dead dye and TCRb antibody for flow detection. Live MC38 cells number in each tube was calculated using beads as an internal control. %Cytotoxicity was calculated as: (MC38blank tube -MC38experimental tube)/MC38experimental tube x 100%.
For the Real-Time Cell Analyzer System (RTCA) based method, 4T1 cells (1 × 104/well) were seeded into 96-well E-plates (ACEA Biosciences) and monitored using the RTCA system in cell incubator for 24 h. CAR-T or control T cells were added at increasing E:T ratios (1:1, 2:1, 4:1, 8:1). Relative cell impedance (cell index) was monitored for 12 h.
For IncuCyte® live cell analysis, MC38 cells were first seeded in 96-well plates (3 × 104/well) for 12 h, after which CAR-T or control T cells were added at increasing E:T ratios (1:8, 1:4, 1:2, or 1:1) at time zero. IncuCyte® Cytotox Red Reagent (Essen BioScience) was then added to the culture medium at a final concentration of 250 nM, and co-culture plates were incubated at 37°C with 5% CO2 and monitored using the IncuCyte system (Essen BioScience) every hour for 24 h. Red fluorescence was normalized to the baseline to quantify dead MC38 cells.
Cytokine ELISA Assay
1 × 104 target cells (4T1, 3T3 or MC38) were cultured with effector cells (CAR-T or control T cells) at increasing E:T ratios (1:1, 2:1, 4:1, 8:1) for 12 h. After centrifuging the plate at 1500 rpm for 5 minutes, supernatant was collected and diluted 10 times, then analyzed by mouse IFN-γ Quantikine ELISA Kit (R&D) according to the manufacturer’s protocol.
For animal studies, mouse serum was collected from the tail vein on day 3 after T cell infusion. Then, 1:10 diluted serum was tested by mouse IFN-γ Quantikine ELISA Kit (R&D) and mouse IL-6 Quantikine ELISA Kit (R&D) according to the manufacturer’s protocol.
Tumor models
For in vivo efficacy experiments, BALB/c mice were inoculated on the right flank with 1 × 105 4T1-luciferase cells and treated with 100 mg/kg CTX on day 5. Two days later, mice were intravenously injected with 3 × 106 CAR-T cells. Tumors were measured twice per week with calipers and tumor sizes were calculated as (length×width2)/2. Mice were intraperitoneally injected with 150 mg/kg XenoLight D-Luciferin, Potassium Salt (PerkinElmer) 15 min before in vivo imaging on day 29 using an IVIS Lumina III in vivo imaging system (PerkinElmer). For the MC38 tumor model, 2 × 106 MC38 cells were subcutaneously injected in the right flank, after which mice were treated with 100 mg/kg CTX on day 23 for preconditioning. Two days later, mice were intravenously injected with 5 × 106 CAR-T cells and tumor sizes were monitored every one or two days.
For in vivo toxicity studies, BALB/c mice were subcutaneously injected with 1 × 105 4T1 cells or PBS on day 0, followed by the indicated numbers of CAR-T or control T cells at the indicated time points. Body weights and animal behaviors were monitored once per day at a fixed time. Mice with 20% or more body weight loss were euthanized according to our approved animal protocol. For the C57BL/6 J model, 1 × 106 cells MC38 colon cancer cells or PBS were intravenously injected into mice, while T cell infusion procedures were identical to those in BALB/c mice.
For mouse serum analysis, 1 × 107 CAR-T or control T cells were transferred into BALB/c mice, and serum was collected from heart on day 3 post T cell infusion. Samples were analyzed by ROCHE COBAS Integra 400Plus/C501 according to the manufacturer’s instruction. All mice were pretreated with 100 mg/kg CTX 2 days before T cell transfer.
T cell in vivo proliferation and accumulation model
EpCAM CAR-modified or untransduced control thy1.1 + T cells (5 × 106) were 1:1 mixed with thy1.2+ untransduced control T cells (5 × 106), after which mixed T cells were intravenously injected into thy1.1+ thy1.2+ B6 mice. Three days later, peripheral blood was subjected to flow cytometry analysis to evaluate proportions of transferred T cells.
EGFRvIII-specific CAR-T cells containing the same intracellular signaling domains as EpCAM CAR-T were used as a control. 1 × 107 thy1.2+ derived EGFRvIII CAR-T cells or EpCAM CAR-T cells were transferred into thy1.1+ B6 mice separately. Mice were euthanized on day 3 post T cell infusion, and spleen, colon, and lung were dissected, minced, and digested to obtain a single-cell suspension. All samples were incubated in ACK lysis buffer on ice for 30 sec. Numbers of transferred and recipient T cells in each mouse were analyzed by flow cytometry. To measure in vivo proliferation, 12 h before flow analysis, mice were intraperitoneally injected with 20 mg/kg 5-ethynyl-2´-deoxyuridine (EdU) (Cayman Chemical). Transferred CAR-T cell proliferation (indicated by EdU staining) was analyzed using a Click-iT Plus kit (Invitrogen™) according to the manufacturer’s instructions.
Real-time quantitative PCR
Genomic DNA from spleen, colon, and lung were extracted using a GenElute Mammalian Genomic DNA Miniprep Kit (Sigma) when mice were sacrificed on day 3 post T cell infusion. Each qPCR reaction was performed with 100–200 ng genomic DNA using PowerUp™ SYBR™ Green Master Mix (Invitrogen). The EpCAM CAR-T MSCV plasmid was diluted and mixed with 100 ng of genomic DNA from mice with no T cell transfer to generate a 6-point standard curve. The lowest limit of quantification (LLOQ) for this assay was determined by the standard curve at 150 copies/100 ng input DNA. Forward primer: 5ʹ-TGC CAG AGA CTT TGC AGC GTA-3ʹ; reverse primer: 5ʹ-CTC TCA GCT CAT AGC CTC CT-3ʹ. The standard cycling mode from the manufacturer’s instruction was used for the qPCR reaction.
Pathological analysis
Mouse lung, intestine, liver, kidney, heart, and spleen were collected and fixed in 4% paraformaldehyde (PFA) solution in PBS for 24 h, then embedded in paraffin. Fresh mouse lung was perfused with PBS before fixation. Five μm sections were cut and stained with hematoxylin and eosin for light microscopy. For immunohistochemical staining, sections were deparaffinized with xylene twice and gradient rehydrated in decreasing concentrations of ethanol, and then sections were washed with PBS 3 times, after which sodium citrate buffer (10 mM, pH 6.0) incubation was used for epitope retrieval (10 min). Peroxidases were inactivated using 3% H2O2 in methanol. 10% goat serum albumin in PBS was used to prevent nonspecific binding (60 min). Next, sections were incubated with anti-mouse EpCAM antibody (ab221552, 1:1000 dilution) and anti-mouse CD3 antibody (ab5690, 1:100 dilution) overnight at 4°C. Slides were washed 3 times with PBS and incubated with goat anti-rabbit HRP polymer (ab214880, 1:1 dilution) for 30 min at 37°C. Slides were developed using a diaminobenzene (DAB) kit (ab64238) at room temperature for 30 sec and rinsed in running water for 5 min. Slides were then counterstained with hematoxylin at room temperature for 1 min. Slides were rinsed in distilled water for 5 min and dehydrated with graded ethanol and xylenes, followed by mounting. Slides were scanned using an Olympus Whole Slide Scanner (VS120). For T cell counting, fields of each section were randomly selected. Intestine sections were reviewed and counted blindly by an independent investigator. Lung sections were analyzed using Imagepro plus 6 (Media Cybernetics, Inc.).
Statistical analysis
Data analysis was conducted as indicated in the figure legends using GraphPad Prism 7 software (GraphPad Software Inc., La Jolla, CA, USA). Welch’s t test was used when the two samples had unequal variances. P < .05 was considered as statistically significant; *P < .05; **P < .01; ***P < .001; ****P < .0001.
Results
In vitro activity of mouse EpCAM-targeted CAR-T cells
To investigate the possible impact of an intact immune system on EpCAM CAR-T therapy, we genetically engineered mouse T lymphocytes with retrovirus comprising a third-generation CAR moiety ().Citation32 This CAR construct encodes a single-chain variable fragment (scFv) derived from a rat anti-mouse EpCAM monoclonal antibody (mAb) G8.8Citation30 followed by a mouse CD8α hinge and transmembrane segment and cytoplasmic signaling domains. The intracellular signal region contains the costimulatory domains of both mouse CD28 and CD137 (4–1BB), followed by the cytoplasmic domain of CD3ζ. To accurately monitor CAR surface expression, we generated a recombinant protein consisting of mouse EpCAM tagged with a human IgG constant fragment (Fc), which can be readily detected by fluorescence-labeled anti-human IgG Fc secondary antibody. As evaluated by flow cytometry, 48 h after retrovirus transduction (), 60% to 95% of T cells were transduced and expressing the CAR structure on their surface.
Figure 1. In vitro activity of mouse EpCAM-targeted CAR-T cells.
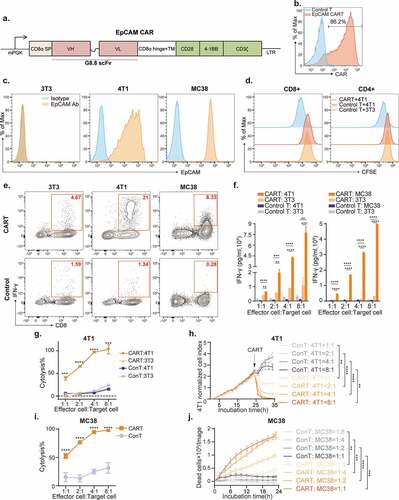
We investigated the in vitro activity of these mouse EpCAM CAR-T cells against various EpCAM positive mouse tumor cell lines. Both the BALB/c derived breast cancer cell line 4T1 and the C57BL/6 J derived colon cancer cell line MC38 express high EpCAM on the cell surface, making these lines ideal target cells. Deficient in EpCAM expression, NIH-3T3 fibroblasts were utilized as a negative control (). We first measured the target-induced proliferation of EpCAM CAR-T cells. For this, CFSE-labeled EpCAM CAR-T cells were co-cultured with 4T1 tumor cells at a 1:1 effector-to-tumor cell (E:T) ratio for 72 h. While untransduced control T cells had almost no proliferative response to EpCAM expressing target cells, both CD4+ and CD8+ subpopulations of CAR-T cells proliferated, as indicated by dilution of CFSE, in response to target cell stimulation ().
Antigen-specific cytokine production in CAR-T cells was examined by both intracellular cytokine staining and ELISA. In order to target the 4T1 and MC38 cell lines, EpCAM CAR was introduced into T cells isolated from BALB/c and C57BL/6 J lymph nodes, respectively, and, co-cultured at an E:T ratio of 1:1 with the corresponding target cells. Interferon-gamma (IFN-γ) production was detected after 12-h co-culture in EpCAM-stimulated CAR-T cells (), whereas IFN-γ secretion was measured at various E:T ratios (1:1,2:1,4:1 and 8:1) after 12-h co-culture. CAR-T cells, but not untransduced control T cells, were activated and secreted IFN-γ in a dose-dependent manner ().
The antigen-specific cytolysis capacity of CAR-T cells was quantified using lactic acid dehydrogenase (LDH) release and real-time cytotoxicity assays (RTCA). BALB/c T cells with or without EpCAM CAR were co-cultured with antigen-positive 4T1 cells or antigen-negative NIH-3T3 cells separately for 12 h, after which we measured LDH released by killed cells. While no toxicity was detected against EpCAM negative NIH-3T3 cells, CAR-T cells executed cytolytic activity against the 4T1 cell line (). RTCA also indicated that EpCAM CAR-T cells effectively kill 4T1 cells in a dose-depended manner (). Similarly, C57BL/6 J derived T cells armed with an EpCAM CAR also specifically and efficiently lysed MC38 colon tumor cells (,j). Together, these in vitro activities validated functionality and specificity of our murine EpCAM CAR are similar to those of previously reported human CAR-T cells.Citation21–Citation23
In vivo efficacy and dose-dependent toxicity of mouse EpCAM-targeted CAR-T cells
We adoptively transferred EpCAM CAR-T cells into syngeneic mice to evaluate their in vivo anti-tumor activity. For this, BALB/c mice were subcutaneously injected with 4T1-luciferase (4T1-LUC) tumor cells on the right flank. Seven days after tumor inoculation, and 2 days post cyclophosphamide (CTX) preconditioning, 3 × 106 EpCAM CAR-T cells or control T cellsCitation33-Citation36 were infused for therapy. EpCAM CAR-T cells reduced tumor burdens to undetectable levels in 4/5 mice on day 29 (). Bioluminescence imaging confirmed that, in comparison with control T cell treatments, tumor burdens in CAR-T-treated mice were significantly diminished (). EpCAM CAR-T cell treatment also impaired MC38 tumor growth in C57BL/6 J mice () and significantly extended animal survival (). These results demonstrate that mouse EpCAM CAR-T cells have in vivo efficacy against syngeneic tumor transplants in both BALB/c and C57BL/6 J mice.
Figure 2. In vivo activity of mouse EpCAM CAR-T cells.
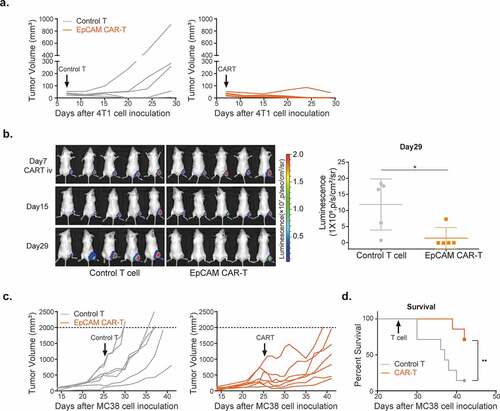
However, during these experiments, we also observed body weight loss of experimental animals which occurred gradually following CTX intraperitoneal injection with peak losses reaching an average of 7.9% (ranging from 2.5% to 14.4%) of maximum body weight. Although at this treatment dosage, animal weights eventually recovered with supportive care provided to mice, the losses were more severe than baseline CTX toxicities observed in the control group (). To investigate this further we increased the dosage of transferred CAR-T cells to 5 × 106, 7 × 106, and 10 × 106 cells/mouse (-d). Accompanying this dose escalation, body weight losses in CAR-T treated mice became more severe, recovery times became longer, and additional toxicity symptoms including decreased activity, hunched posture, and ruffled fur were observed. Critically, 5 of 9 mice died or were over the 20% weight loss limit in the 10 × 106 CAR-T cells treatment group, which reached the humane point for experiment termination (, right panel). In contrast, using the same dose escalation, no significant toxicity was observed in control T cell-treated mice. Taken together, these data demonstrate that irreversible toxicity was not a consequence of CTX pre-conditioning, but rather of EpCAM CAR-T cell infusion.
Figure 3. Mouse EpCAM CAR-T cells induce acute dose-dependent toxicities in both tumor-bearing and tumor-free mice.
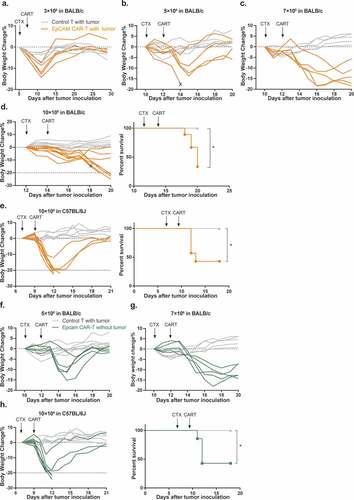
To examine whether this severe toxicity is strain-specific, we performed similar CAR-T treatment in C57BL/6 J mice implanted with 1 × 106 EpCAM positive MC38 mouse colon cancer cells. We again observed that infusion of 10 × 106 EpCAM CAR-T cells led to animal mortality (57.1%, ). Toxicity symptoms became evident in C57BL/6 J mice on day 3 post infusion, and, were essentially the same as those observed in BALB/c mice.
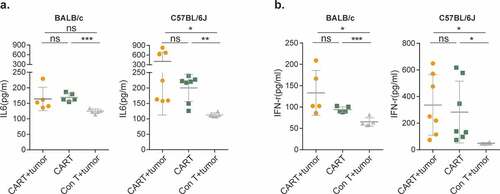
To distinguish whether CAR-T-produced mortality results from CRS or on-target/off-tumor toxicity, we infused increasing doses of EpCAM CAR-T cells into tumor-free BALB/c and C57BL/6 J mice. Like their tumor-bearing counterparts, non-tumor-bearing mice also displayed dose-dependent clinical symptoms, body weight loss, and even lethality (-h). Upon stimulation with cognate antigen, CAR-T cells release inflammatory cytokines and chemokines, including IFN-γ and tumor-necrosis factor α (TNF-α), as well as IL6, IL1, and IL10 to trigger monocyte and macrophage activation, thereby initiating CRS.Citation11,Citation12 Thus, we analyzed the levels of two major cytokines, IL-6 () and IFN-γ (), present in sera from CAR-T treated BALB/c and C57BL/6 J mice. In comparison with tumor-bearing mice treated with control T cells, both cytokines were significantly elevated in both tumor-carrying and tumor-free CAR-T cell-treated cohorts. Together, these results strongly suggested that murine EpCAM CAR recognizes and is activated by EpCAM antigen expressed on non-tumor tissues, resulting in on-target/off-tumor toxicity.Citation8,Citation9,Citation37-Citation39
Figure 4. EpCAM CAR-T-mediated CRS is tumor independent.
EpCAM CAR-T cells attack normal lung tissue
We next performed pathological studies in order to identify target organs recognized by EpCAM CAR-T cells. Among healthy tissues, EpCAM has the highest expression in mouse () and human intestine.Citation40,Citation41 Also, due to the significant body weight loss observed in mice after CAR-T cell infusion, we hypothesized that intestine might be the primary target of EpCAM CAR-T cells such that damage to the digestive system is the major toxicity. However, histopathological analyses of tissues collected from CAR-T treated and moribund mice revealed no obvious pathological changes in mouse intestine (supplement Figure 1a), with no bleeding or ulcers seen by visual inspection. Moreover, Immunohistochemistry (IHC) staining showed intact mucosa and villi with no indication of abnormal lymphocyte infiltration ().
Figure 5. Alveolar cells express EpCAM and are targeted by EpCAM CAR-T cells.
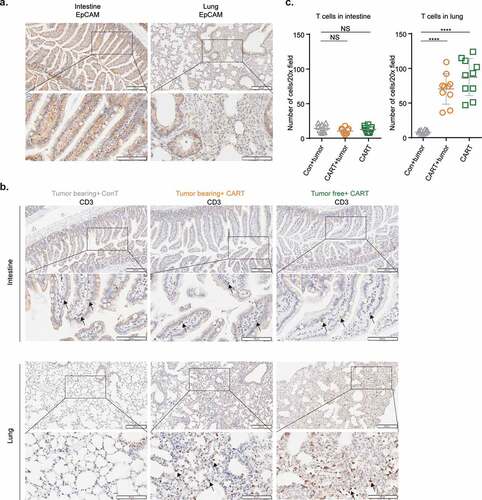
Histopathological screening of other major organs such as heart, liver, spleen, and kidney (supplement Figure 1b) also failed to detect any significant pathological changes after CAR-T treatment regardless of tumor-bearing status. Moreover, mouse serum biochemical testing for markers indicative of liver, pancreas, heart, and kidney damage revealed no significant differences between mice treated with EpCAM CAR-T and control T cells (supplement Figure 2).
Because intravenously injected CAR-T cells are known to first reach and temporally reside in the lung,Citation42 we examined whether EpCAM is expressed in lung tissue. Like the expression pattern observed in mouse intestine, strong EpCAM expression was identified in lung bronchioles, where it is mainly restricted to luminal epithelial cells (). However, like in intestine, this EpCAM does not cause lymphocyte accumulation near bronchioles, which remain intact (). Nevertheless, we observed other severe pathological responses in lung tissue, including congestion (supplement Figure 1c), diffuse thickening of the alveolar septum, alveolar structural deterioration, and inflammatory cell infiltration (supplement Figure 1d). Within the lung, besides bronchial epithelial cells, EpCAM is also expressed on the surface of lung alveolar cells, albeit at a lower level (). Accordingly, we detected massive T cell infiltrated in CAR-T treated lung tissues (, lower panel and ). These findings suggest that alveolar EpCAM expression in normal lung recruits EpCAM CAR-T cells, resulting in CAR-T activation, lung inflammation, and eventual tissue damage.
Antigen-dependent and selective accumulation of EpCAM CAR-T cells in lung
To quantitatively assess the selective tissue distribution of infused EpCAM CAR-T cells, we performed transfer experiments with tumor-free mice. For this, we mixed congenitally labeled control effector T cells (Thy1.2) and EpCAM CAR-T cells (Thy1.1) at a 1:1 ratio and transferred them into Thy1.1+ Thy1.2+ recipients. Three days after infusion, flow cytometry analysis showed that in mouse peripheral blood the ratio between control T cells and EpCAM CAR-T cells was on average 2.92:1 (), indicating that approximately two-thirds (65.8%) of EpCAM CAR-T cells were absent from the circulation.
Figure 6. Antigen-dependent and selective accumulation of EpCAM CAR-T cells in lung.
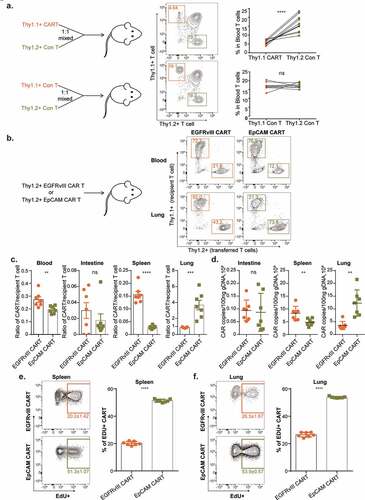
To reveal the antigen-dependency of EpCAM CAR-T distribution, we employed an antigen-irrelevant CAR against epidermal growth factor receptor variant III (EGFRvIII) as a reference.Citation31 EGFRvIII is a tumor-specific antigen (TSA) generated by a tumor-initiating in-frame deletion of exons 2–7 in glioblastoma cells, such that the EGFRvIII antigen is absent from normal tissues.Citation43 Excluding the antigen-recognizing scFv segment, the molecular structures of our EpCAM and EGFRvIII CARs are identical (supplement Figure 3a). To assess CAR-T cell localization, 1 × 107 thy1.2+ EGFRvIII CAR-T cells or EpCAM CAR-T cells were transferred into thy1.1+ recipient mice separately. Whereas EpCAM CAR-T treatment caused significant body weight loss, body weights of mice in the EGFRvIII CAR-T treatment group remained stable 3 days post T cell infusion (supplement Figure 3b). Flow analysis conducted on day 3 further showed that, compared with EGFRvIII CAR-T cells, EpCAM CAR-T cell numbers were significantly reduced in blood and spleen but selectively accumulated in the lung (-c). We next performed qPCR-based molecular quantification in order to validate this FACS analysis (supplement Figure 3c). Genomic DNA was extracted from various tissues and CAR gene copy numbers were measured, confirming that both EGFRvIII and EpCAM CAR-T cells have a low presence in the mouse intestine. In contrast, utilizing EGFRvIII CAR as a standard, EpCAM CAR-T cells accumulate in lung tissue with a concomitant reduction in the spleen ().
To further demonstrate the impact of cognate antigen on CAR-T cells, taking proliferation as the parameter, we measured CAR-T cell activation in tumor-free mice. Sixty hours post infusion, EdU was intraperitoneally administered to the mice, after which flow analysis was conducted using T cells from spleen and lung collected 12 h post EdU labeling. CAR-T cells were generated in vitro through anti-CD3 and anti-CD28 stimulated proliferation. Therefore, we observed 20.2% and 26.5% of EGFRvIII CAR-T cells were labeled with EdU in the spleen and lung, respectively. This represents the baseline for remaining CAR-T expansion potential 3 days post transfer in the absence of cognate antigen. However, for EpCAM CAR-T cells, 51.3% and 53.9% cells were EdU positive ( and ). Based on these results, we conclude that EpCAM CAR-T cells are activated to proliferate in response to encountering basal EpCAM expressed in tumor-free mice.
Discussion
Due to inspiring clinical results demonstrating robust anti-CD19 CAR-T therapeutic efficacy against hematological malignancies, many attempts have also been made to apply CAR-T cell therapies for solid tumor treatment. While surface-displayed TSAs are rare, the current focus is on identifying tumor-associated antigens (TAAs), which are heavily expressed on tumor cells but have limited presence in normal tissues. Due to this basal expression, the major concern for targeting TAAs is the possibility of on-target/off-tumor side effects, which may lead to lethality in patients.Citation8,Citation10,Citation29,Citation44 With this caution, EpCAM was considered to be one of the most ideal TAA target: it is highly expressed on the surface of a variety of epithelial-derived tumor cells;Citation45 and, although it can be found on the membrane of normal simple epithelia, it is expressed at lower levels and, most importantly, limited to the basolateral cell membrane or lateral interfaces of polarized epithelial cells. The assumption is that, unlike its universal display on the tumor cell surface, this restricted pattern of expression may limit the access of EpCAM-specific CAR-T cells in normal tissues, thereby mitigating on-target/off-tumor effects.
On the one hand, this is indeed the case in our observations: murine EpCAM-specific CAR-T cells failed to act on intestinal EpCAM, even though this molecule is highly expressed from crypts to villi. This indicates that the polarized and restricted expression of EpCAM shields this molecule from CAR-T access. On the other hand, while EpCAM expression is rather weak on the surface of lung alveolar cells, it is sufficient to attract and activate CAR-T cells. We speculate that tissue anatomy may play a major role in this discrepancy. Within the GI tract, gut endothelial cells are connected by tight or adherent junctions, which are further enclosed by enteric glial cells. Together, this organization comprises the gut–vascular barrier (GVB).Citation46–Citation48 The GVB prevents bacteria within the gut from entering the bloodstream, while similarly impeding circulating EpCAM CAR-T cells from reaching the basal epithelia. In contrast, within the lung, alveolar cells are in close proximity (<0.5 μm in distance) to the capillary to form an alveolar-capillary barrier.Citation49 This enables oxygen and carbon dioxide exchange, but also provides direct access for EpCAM CAR-T cells to reach their unwanted targets, and is similar to the fatal case reported by Morgan et al.Citation10 There, low basal expression of the HER2 antigen on lung epithelial cells was shown to activate HER2-targeting CAR-T cells when infused at a high-dose (109 cells), leading to inflammatory cytokine secretion and lethal toxicity. However, this toxicity was not observed after the administration of lower doses of HER2 CAR-T cells,Citation50 highlighting the importance of optimizing CAR-T dosing protocols.
For our EpCAM CAR, the severe toxicity was observed in recipient mice on both BALB/c and C57BL/6 genetic backgrounds. It is well known that T cell effector responses are biased to Th2 type in BALB/c mice, whereas to Th1 type in C57BL/6 mice.Citation51,Citation52 Despite these differences, our murine EpCAM CAR-T cells exhibited comparable levels of lethal lung toxicity with no obvious differences in severity. This further supports our belief that mouse lung injury is primarily caused by direct the on-target-off-tumor CAR-T cytolysis, while biased T cell differentiation plays a limited role.
In conclusion, the current study highlights the need for conducting proper preclinical toxicity analysis prior to moving new CAR-T therapies into the clinic. The dose-dependent, life-threatening lung toxicity observed here has not been reported in preclinical models infusing human EpCAM CAR-T into immunodeficient mice. While several clinical trials examining EpCAM CAR-T cells are currently recruiting patients, our results advise investigators to revisit the possibility of severe off-tumor toxicities. To utilize EpCAM as a viable target, treatment strategies will need to be optimized for human cancer; these may include: 1) determining a suitable dosage window with minimal toxicity and sufficient anti-tumor efficiency; 2) developing a distribution-restricted CAR-T that eradicates tumors locally; and, 3) modifying the scFv affinity, such that CAR-T cells can only be activated by the high density of EpCAM protein which exists on the surface of tumor cells, but not on lung alveoli.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Conflicts of interest
Qi-Jing Li is a scientific co-founder and shareholder of TCRCure Biopharma.
Supplemental Material
Download ()Acknowledgments
We would like to thank the staff of the mouse facility and flow cytometry facility of Duke University. The MC38 cell line was kindly provided by the National Cancer Institute.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–11. doi:10.1056/NEJMoa1407222.
- Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi:10.1016/S0140-6736(14)61403-3.
- Park JH, Geyer MB, Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood. 2016;127:3312–3320. doi:10.1182/blood-2016-02-629063.
- Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, et al. CD19 CAR-T cells of defined CD4+: CD8+composition in adult B cell ALL patients. J Clin Invest. 2016;126:2123–2138. doi:10.1172/JCI85309.
- Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, Bleakley M, Brown C, Mgebroff S, Kelly-Spratt KS, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129:3322–3331. doi:10.1182/blood-2017-02-769208.
- Administration TUSFaD. FDA approves CAR-T cell therapy to treat adults with certain types of large B-cell lymphoma. The U.S. Food and Drug Administration: The U.S. Food and Drug Administration; 2017.
- Administration TUSFaD. FDA approves tisagenlecleucel for adults with relapsed or refractory large B-cell lymphoma. The U.S. Food and Drug Administration: The U.S. Food and Drug Administration; 2018.
- Lamers CH, Klaver Y, Gratama JW, Sleijfer S, Debets R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem Soc Trans. 2016;44:951–959. doi:10.1042/BST20160037.
- Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, Vulto A, den Bakker M, Oosterwijk E, Debets R, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21:904–912. doi:10.1038/mt.2013.17.
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18:843–851.
- Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, Sanvito F, Ponzoni M, Doglioni C, Cristofori P, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24:739–748. doi:10.1038/s41591-018-0036-4.
- Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018;24:731–738. doi:10.1038/s41591-018-0041-7.
- Wegner A. Chimeric antigen receptor T cells for the treatment of cancer and the future of preclinical models for predicting their toxicities. Immunotherapy. 2017;9:669–680. doi:10.2217/imt-2017-0028.
- Siegler EL, Wang P. Preclinical Models in Chimeric Antigen Receptor-Engineered T-Cell Therapy. Hum Gene Ther. 2018;29:534–546. doi:10.1089/hum.2017.243.
- Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, Watanabe T, Akashi K, Shultz LD, Harada M, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood. 2005;106:1565–1573. doi:10.1182/blood-2005-02-0516.
- Leonard WJ. Cytokines and immunodeficiency diseases. Nat Rev Immunol. 2001;1:200–208. doi:10.1038/35105066.
- Kovanen PE, Leonard WJ. Cytokines and immunodeficiency diseases: critical roles of the gamma(c)-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol Rev. 2004;202:67–83. doi:10.1111/j.0105-2896.2004.00203.x.
- Trzpis M, McLaughlin PM, de Leij LM, Harmsen MC. Epithelial cell adhesion molecule: more than a carcinoma marker and adhesion molecule. Am J Pathol. 2007;171:386–395. doi:10.2353/ajpath.2007.070152.
- Xie X, Wang CY, Cao YX, Wang W, Zhuang R, Chen LH, Dang NN, Fang L, Jin BQ. Expression pattern of epithelial cell adhesion molecule on normal and malignant colon tissues. World J Gastroenterol. 2005;11:344–347. doi:10.3748/wjg.v11.i3.344.
- Spizzo G, Fong D, Wurm M, Ensinger C, Obrist P, Hofer C, Mazzoleni G, Gastl G, Went P. EpCAM expression in primary tumour tissues and metastases: an immunohistochemical analysis. J Clin Pathol. 2011;64:415–420. doi:10.1136/jcp.2011.090274.
- Ang WX, Li Z, Chi Z, Du SH, Chen C, Tay JC, Toh HC, Connolly JE, Xu XH, Wang S, et al. Intraperitoneal immunotherapy with T cells stably and transiently expressing anti-EpCAM CAR in xenograft models of peritoneal carcinomatosis. Oncotarget. 2017;8:13545–13559. doi:10.18632/oncotarget.14592.
- Deng Z, Wu Y, Ma W, Zhang S, Zhang YQ. Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol. 2015;16:1. doi:10.1186/s12865-014-0064-x.
- Zhang BL, Li D, Gong YL, Huang Y, Qin DY, Jiang L, Liang X, Yang X, Gou H-F, Wang Y-S, et al. Preclinical Evaluation of Chimeric Antigen Receptor-Modified T Cells Specific to Epithelial Cell Adhesion Molecule for Treating Colorectal Cancer. Hum Gene Ther. 2019;30:402–412. doi:10.1089/hum.2018.229.
- EpCAM CAR-T for Treatment of Nasopharyngeal Carcinoma and Breast Cancer. [accessed 2019 Sep 30]. ClinicalTrials.gov: ClinicalTrials.gov
- Intraperitoneal Infusion of EpCAM CAR-T Cell in Advanced Gastric Cancer With Peritoneal Metastasis (WCH-GC-CART). [accessed 2019 Sep 30]. ClinicalTrials.gov: ClinicalTrials.gov
- A Clinical Research of CAR T Cells Targeting EpCAM Positive Cancer (CARTEPC). [accessed 2019 Sep 30]. ClinicalTrials.gov: ClinicalTrials.gov
- Study Evaluating the Efficacy and Safety With CAR-T for Liver Cancer (EECLC). [accessed 2019 Sep 30]. ClinicalTrials.gov: ClinicalTrials.gov
- Study Evaluating the Efficacy and Safety With CAR-T for Stomach Cancer (EECSC). [accessed 2019 Sep 30]. ClinicalTrials.gov: ClinicalTrials.gov
- Gross G, Eshhar Z. Therapeutic Potential of T Cell Chimeric Antigen Receptors (CARs) in Cancer Treatment: counteracting Off-Tumor Toxicities for Safe CAR T Cell Therapy. Annu Rev Pharmacol Toxicol. 2016;56:59–83. doi:10.1146/annurev-pharmtox-010814-124844.
- Farr A, Nelson A, Truex J, Hosier S. Epithelial heterogeneity in the murine thymus: a cell surface glycoprotein expressed by subcapsular and medullary epithelium. J Histochem Cytochem. 1991;39:645–653. doi:10.1177/39.5.2016514.
- Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, Schmittling RJ, Nair SK, Reap EA, Norberg PK, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. 2014;20:972–984. doi:10.1158/1078-0432.CCR-13-0709.
- Hartmann J, Schussler-Lenz M, Bondanza A, Buchholz CJ. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med. 2017;9:1183–1197. doi:10.15252/emmm.201607485.
- Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR T Cells Expressing IL-12 Eradicate Lymphoma in Fully Lymphoreplete Mice through Induction of Host Immunity. Mol Ther Oncolytics. 2018;8:41–51. doi:10.1016/j.omto.2017.12.003.
- Wada S, Yoshimura K, Hipkiss EL, Harris TJ, Yen HR, Goldberg MV, Grosso JF, Getnet D, Demarzo AM, Netto GJ, et al. Cyclophosphamide augments antitumor immunity: studies in an autochthonous prostate cancer model. Cancer Res. 2009;69:4309–4318. doi:10.1158/0008-5472.CAN-08-4102.
- Cheadle EJ, Hawkins RE, Batha H, Rothwell DG, Ashton G, Gilham DE. Eradication of established B-cell lymphoma by CD19-specific murine T cells is dependent on host lymphopenic environment and can be mediated by CD4+ and CD8+ T cells. J Immunother. 2009;32:207–218. doi:10.1097/CJI.0b013e318194a921.
- Lowe KL, Mackall CL, Norry E, Amado R, Jakobsen BK, Binder G. Fludarabine and neurotoxicity in engineered T-cell therapy. Gene Ther. 2018;25:176–191. doi:10.1038/s41434-018-0019-6.
- Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–2. doi:10.1200/JCO.2006.05.9964.
- Hombach A, Hombach AA, Abken H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010;17:1206–1213. doi:10.1038/gt.2010.91.
- Drent E, Themeli M, Poels R, de Jong-korlaar R, Yuan H, de Bruijn J, Martens ACM, Zweegman S, van de Donk NWCJ, Groen RWJ, et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol Ther. 2017;25:1946–1958. doi:10.1016/j.ymthe.2017.04.024.
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015 Jan 23;347(6220):1260419. doi:10.1126/science.1260419.
- Bastian F, Parmentier G, Roux J, Moretti S, Laudet V, Robinson-Rechavi M. Mouse tissue expression of EpCAM. Mouse tissue expression of EpCAM: Bgee. New York (NY): Springer Nature; 2008.
- Visioni A, Kim M, Wilfong C, Blum A, Powers C, Fisher D, Gabriel E, Skitzki J. Intra-arterial Versus Intravenous Adoptive Cell Therapy in a Mouse Tumor Model. J Immunother. 2018;41:313–318. doi:10.1097/CJI.0000000000000235.
- Gan HK, Cvrljevic AN, Johns TG. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. Febs J. 2013;280:5350–5370. doi:10.1111/febs.12393.
- Sun S, Hao H, Yang G, Zhang Y, Fu Y. Immunotherapy with CAR-Modified T Cells: toxicities and Overcoming Strategies. J Immunol Res. 2018;2018:2386187. doi:10.1155/2018/2386187.
- Went PT, Lugli A, Meier S, Bundi M, Mirlacher M, Sauter G, Dirnhofer S. Frequent EpCam protein expression in human carcinomas. Hum Pathol. 2004;35:122–128. doi:10.1016/j.humpath.2003.08.026.
- Gentile ME, King IL. Blood and guts: the intestinal vasculature during health and helminth infection. PLoS Pathog. 2018;14:e1007045. doi:10.1371/journal.ppat.1007045.
- Ungaro F, Tacconi C, D’Alessio S. Beyond Intestinal Barrier: the Blood Endothelium as a Second Wall of Defense Against Bacterial Invasion. Gastroenterology. 2016;150:1678–1680. doi:10.1053/j.gastro.2016.04.024.
- Spadoni I, Zagato E, Bertocchi A, Paolinelli R, Hot E, Di Sabatino A, Caprioli F, Bottiglieri L, Oldani A, Viale G, et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science (New York, NY). 2015;350:830–834. doi:10.1126/science.aad0135.
- Barrowcliffe MP, Jones JG. Solute permeability of the alveolar capillary barrier. Thorax. 1987;42:1–10. doi:10.1136/thx.42.1.1.
- Bernhard H, Neudorfer J, Gebhard K, Conrad H, Hermann C, Nährig J, Fend F, Weber W, Busch DH, Peschel C, et al. Adoptive transfer of autologous, HER2-specific, cytotoxic T lymphocytes for the treatment of HER2-overexpressing breast cancer. Cancer Immunology, Immunotherapy: CII. 2008;57:271–280. doi:10.1007/s00262-007-0355-7.
- Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166–6173. doi:10.4049/jimmunol.164.12.6166.
- Hsieh CS, Macatonia SE, O’Garra A, Murphy KM. T cell genetic background determines default T helper phenotype development in vitro. J Exp Med. 1995;181:713–721. doi:10.1084/jem.181.2.713.