
ABSTRACT
Therapeutic blockade of PD-1/PD-L1 signaling with monoclonal antibodies (mAbs) has shown clinical success and activity across a broad set of cancer subtypes. However, monotherapy with PD-1/PD-L1 inhibitors are only effective in a subset of patients and ongoing studies show efficacy of treatment depends on a combinatorial approach. Contrary to mAbs chimeric B-cell cancer vaccines incorporating a “promiscuous” T-cell epitope have the advantage of producing a polyclonal B-cell antibody that can potentially induce memory B- and T-cell responses, while reducing immune evasion and suppression. Here, we describe a novel PD-1 B-cell peptide epitope vaccine (amino acid 92–110; PD1-Vaxx) linked to a measles virus fusion peptide (MVF) amino acid 288–302 via a four amino acid residue (GPSL) emulsified in Montanide ISA 720VG that aims to induce the production of polyclonal antibodies that block PD-1 signaling and thus trigger anticancer effects similar to nivolumab. In preclinical studies, the PD1-Vaxx outperformed the standard anti-mouse PD-1 antibody (mAb 29F.1A12) in a mouse model of human HER-2 expressing colon carcinoma. Furthermore, the combination of PD1-Vaxx with combo HER-2 peptide vaccine (B-Vaxx) showed enhanced inhibition of tumor growth in colon carcinoma BALB/c model challenged with CT26/HER-2 cells. The PD-1 or combined vaccines were safe with no evidence of toxicity or autoimmunity.
Introduction
One of the most exciting recent developments in cancer immunotherapy has been the introduction of the immune checkpoint blockade, for which J.P. Alison and T. Honjo received the 2018 Nobel Prize in Physiology and Medicine.Citation1 Indeed, therapeutic blockade of the signaling axis between programmed cell death protein-1 (PD-1) and its ligand programmed cell death ligand-1 (PD-L1) with monoclonal antibodies (mAbs) such as the humanized mAb pembrolizumab (Keytruda®) and the fully human mAb nivolumab (Opdivo®) has shown remarkable clinical success in some cancer patients.Citation2,Citation3 Such monotherapies have demonstrated impressive activity across a broad set of cancer subtypes, even at advanced and metastatic stages of disease.Citation4–9 However, 60–70% of patients receiving anti-PD-1 therapy, such as pembrolizumab or nivolumab as single agents remain resistant to this therapy and will not respond or will relapse, leaving a substantial unmet medical need.Citation10 Combinations of checkpoint-blocking antibodies are being tested in clinical trials, which include standard therapies (e.g. chemotherapy, radiation therapy), targeted therapies, other immunologic modulators, tumor vaccines, and oncolytic viruses. Combinations of checkpoint-blocking antibodies are more efficacious than single inhibitors but also cause greater immune-related toxicities (see review in Yan et al.,Citation10). Therefore, the promise of immunotherapy has been tempered by a number of clinical setbacks and is awaiting novel approaches.
Over the past two decades, another cancer treatment paradigm has evolved: selective, mechanism-based targeted therapies using peptide-based B-cell epitopes and peptide immuno-therapeutics that block the growth and spread of cancer by inhibiting oncogenic signaling pathways; these targeted therapies tend to have fewer side effects than nonspecific chemotherapies (reviewed in Kaumaya et al.,Citation11). The human epidermal growth factor receptor 2 (HER-2) also known as HER2, HER-2/neu, p185neu, ERBB2, or CD340 family of receptors plays a central role in the pathogenesis of several human cancers including breast, ovarian, renal, colon, and lung carcinoma cancersCitation12–17 and is associated with more aggressive forms of cancer,Citation18 increased risk of metastasis, increased tumor invasion and decreased overall survival.Citation19,Citation20 Therefore, HER-2 is a key therapeutic target in several cancers that are binding sites of trastuzumab (Herceptin®) and pertuzumab (Perjeta®). Trastuzumab was the first humanized mAb targeting HER-2 in combination with chemotherapy to be approved for clinical use in patients with metastatic HER-2 positive breast cancer.Citation21–28 The addition of pertuzumab, a humanized mAb that blocks the dimerization of HER-2 with other HER family members, to docetaxel and trastuzumab in patients with metastatic HER-2 positive breast cancer, resulted in improvement in progression-free survival from 12.4 months to 18.5 months and in overall survival from 40.8 to 56.5 months.Citation29–31 Despite the benefit observed from trastuzumab approximately one-third of patients with metastatic, HER-2 positive breast cancer initially respond and the majority of responding patients eventually develop acquired resistance within one year of therapy.Citation32 Targeted therapies with humanized mAb to HER-2 (trastuzumab, pertuzumab) or the chimeric mAb specific targeting EGFR (cetuximab, Erbitux®) have markedly improved survival in the adjuvant setting but still demonstrate toxicities, intrinsic or acquired resistance, and the majority of patients with advanced cancer finally succumb to their disease.
Despite their proven utility, mAbs have specific drawbacks as therapeutics when infused in patients, including poor tissue/tumor penetrance, which may be especially pertinent when targeting the PD-1/PD-L1 signaling pathway. Additional drawbacks include immune-related toxicities, inadequate pharmacokinetics (PK) such as binding affinities, specificities, half-life, and high-cost burden to manufacturers and consumers.Citation33 Therefore, in recent years, peptide-based and small-molecule inhibitors have been proposed in drug development strategies based on the publication of the human PD-1/PD-L1 complex in 2015 by Zak et al.,Citation34 However, the development of small-molecule immune checkpoint inhibitors lags far behind mAbs. Although a few peptide-based and small-molecule inhibitors have been reported primarily in patent applications, no further information is disclosed so far.Citation35,Citation36 Importantly, such peptide vaccines offer an attractive immunotherapeutic option in the treatment of cancer, with considerable advantages in safety, cost-effectiveness, and ease of administration.
We have advanced a new paradigm in immunotherapy that focuses on humoral responses based on conformational B-cell epitope vaccines, with the goal of circumventing intrinsic drug resistance whilst providing the possibility of a durable cure. Specifically, our laboratory has developed two novel B-cell epitope-specific vaccines (B-Vaxx): a trastuzumab-binding epitope and the pertuzumab-binding epitopeCitation37,Citation38 designed specifically using the X-ray structures of the (HER-2)-trastuzumab and (HER-2)-pertuzumab complexes.Citation39–41 The clinical results of the first-in-human, dose escalation portion of the Phase I study with B-Vaxx were recently reported.Citation42 The study vaccine was safe, well tolerated, exhibited anti-tumor activity, and showed preliminary indication that peptide vaccination may avoid therapeutic resistance and offer a promising alternative to mAb therapies.
In this report, we describe for the first time the development and characterization of a novel chimeric B-cell peptide epitope targeting human PD-1 linked to measles fusion protein (MVF, sequence 288–302) T-cell epitope which aims to induce the body to produce polyclonal antibodies that block PD-1 signaling, thus mimicking the effects of nivolumab. To test the concept of developing a PD-1 vaccine, we chose the well-established murine colon carcinoma cell line CT26 tumor model in syngeneic BALB/c mice to evaluate the effects of vaccination of four potential PD-1 vaccine candidates. We identified one PD-1 epitope sequence 92–110 epitope (PD1-Vaxx) that significantly reduced tumor growth in a syngeneic BALB/c CT26 model. Importantly, we tested the power of combining the PD1-Vaxx with an established cancer vaccine (B-Vaxx) focusing on a relevant animal model with high translational potential. We obtained enhanced inhibition of tumor growth in a syngeneic BALB/c model challenged with colon carcinoma cell line CT26/HER-2 which outperformed the standard anti-mouse PD-1 mAb (29F.1A12).Citation43
Materials and methods
Identification of hPD-1 peptide epitopes
The selection of candidate B-cell epitopes expressed on the surface of PD-1 was accomplished by an in-house (Peptide CompanionTM, www.5z.com/csps/comer/pcom) computer-aided analysis using six correlates of antigenicity,Citation44 described as follows: (a) the profiles of chain flexibility and mobility of individual sequences were calculated according to Karplus and Schultz;Citation45 (b) hydropathy profiles were generated over a seven residue span setting and then smoothed with a three residue span using the scale of Kyte and Doolittle;Citation46 (c) hydrophilicity profiles over a six residue window were generated using the program of Hopp and Woods;Citation47 (d) analysis of the exposure of an amino acid residue to water (1.4A probe) was carried out by the solvent exposure algorithm of Rose et al.,Citation48; (e) protrusion indices were calculated by the method of Thornton et al. that predicts portions of proteins that are accessible and protrude into the solvent;Citation49 (f) the probability that a five-residue sequence is antigenic was determined by the method of Welling et al.,Citation50sequences were given a score of 1 to 6 based on their respective index values and were ranked: the highest ranking sequences had the highest individual score for the analyses examined, and successive candidates had the next highest score.
The best scoring epitopes were further ranked by correlation with their secondary structural attributes; e.g., an amphiphilic α-helical sequence or a β-turn loop regions are preferred over random coil fragments. Computer programs by Chou and FasmanCitation51 and Novotny et al.,Citation52 were used to predict the secondary structure (α-helix, β-strand/sheet, β-turn/loop, random coil) and α-helical amphiphilic moment. Finally, consideration was given to the individual amino acid sequence. Electrostatic ion pairs and helix dipole interaction in helical segment were also considered (e.g., hydrophobic/hydrophilic balance). We used peptide epitope mapping using algorithms of immunogenicity/antigenicity to identify four epitopes of PD-1 and we combined analysis of these epitopes with crystal structures complex of human PD-1/human PD-L1 (hPD-1/hPD-L1) as disclosed by Zak et al.,Citation34 (PDB ID: 4ZQK) to engineer a chimeric B-cell vaccine based on the extracellular domain of PD-1. The selection was further enhanced by examining the 3-D structure of PD-1Citation53 (PDB ID code: 4Z18, 4ZQK, 3BIK). We have modeled all the four epitopes using PyMOL 3-D modeling software Version 2.4.0 (Schrodinger, New York, NY, USA). The PyMOL User’s Manual: https://pymol.org.
Synthesis of hPD-1 peptide epitopes
Four novel peptide sequences that were identified to target human PD-1 were synthesized using a 9600 Milligen/Biosearch solid-phase peptide synthesizer (Millipore, Bedford, MA, USA) with Fmoc/t-Butyl chemistry and PyBOP/6Cl-HOBT coupling reagents on CLEAR amide resin (Peptides International, Louisville, KY, USA). Some peptide samples were acetylated using 1-Acetylimidazole (Sigma-Aldrich St. Louis, MO, USA) before cleavage to provide samples of peptides with more stable configuration as compared to free peptides which display flexible conformation. All peptides were synthesized as chimeric constructs with a “promiscuous” T helper epitope derived from the measles virus fusion protein (MVF, amino acids 288–302) using a four-residue linker (GPSL). Peptides were cleaved from the resin using cleavage reagent R (TFA)/thioanisole/EDT/anisole (90/5/3/2), and crude peptides were purified by semi preparative (C-4 Vydac columns) reversed-phase-high performance liquid chromatography (RP-HPLC; Waters, Bedford, MA, USA). RP-HPLC fractions showing the same retention time were pooled together and lyophilized. All peptides showed purity in excess of 95%. Samples were then characterized by MALDI (Matrix-Assisted Laser Desorption Ionization) mass spectroscopy at the CCIC (Campus Chemical Instrumentation Center, The Ohio State University, Columbus, OH, USA) and analyzed on an analytical RP-HPLC system (Waters, Bedford, MA, USA). All peptides had the correct molecular mass.
HER-2 peptides mimicking trastuzumab- and pertuzumab-binding sites
The rationale, selection, and synthesis of the two peptide constructs were originally described by the Kaumaya laboratory.Citation37,Citation38 The GMP peptides trastuzumab-binding epitope conformational B-cell epitope (597–626) and the pertuzumab-binding conformational B-cell epitope (266–296) were purchased from Peptisynthia (Torrance, CA) and acquired by Solway Group (Zug, Switzerland). The GMP peptides met all the FDA and US Pharmacopoeia requirements for sterility (i.e. bacterial/fungal), endotoxins, and potency. The bulk peptides were supplied to University of IOWA Pharmaceuticals manufacturing facility (Iowa City, Iowa) for sterile vialing in 3 mg lots. Endotoxin levels of these peptides were tested and determined to be within acceptable levels as Good Manufacturing Practice (GMP) grade. A combination of two HER-2 B-cell epitope (B-Vaxx) successfully completed a Phase I active immunotherapy clinical trial in 2019Citation42 (NCT01376505; IND #14633 2019) and presently undergoing an efficacy trial in HER-2 positive breast and colon cancers.
Specificity of PD-1 peptide binding to rhPD-L1 and nivolumab by surface plasmon resonance (SPR)
The specificity was determined by SPR spectroscopy (Biacore T200, Columbia, MD) at 25°C and binding affinities to immobilized recombinant human PD-L1 (rhPD-L1) purchased from (Acrobiosystems, Inc, Newark, DE) and nivolumab (obtained from the OSU James Pharmacy, Columbus, OH) on CM5 sensor chip (GE Healthcare Bio-Sciences, Uppsala, Sweden) were determined. rhPD-L1 ectodomain was immobilized onto the gold surface of a CM5 sensor chip by direct amine coupling. To obtain theoretical maximum response upon peptide binding, we calculated immobilization amount of rhPD-L1, nivolumab, and human IgG: isotype control human IgG isotype control (ThermoFisher, Rockford, IL) is 9790 RU, 14286 RU, and 14286 RU, respectively. 20 μg/ml of rhPD-L1 at 10 mM NaAc pH 5.5, nivolumab at 10 mM HEPES, pH 7.5 and human IgG at 10 mM HEPES, pH 7.0 was injected over chip after activation with EDC/NHS for 7 min at 10 μl/min. The resulting immobilization levels for rhPD-L1, nivolumab and human IgG were 2345 RU, 12264 RU and 11651 RU, respectively. To validate prepared sensor chip, 1 μM (17.3 μg/ml) rhPD-1 was injected over the chip for 3 min at 10 μl/min (data not shown). 1 μM BSA was used as the negative control. The chip was regenerated by 10 mM glycine-HCl, pH 2.5
Immunization with MVF hPD-1 peptide epitopes
For each peptide, vaccine antibodies were raised using New Zealand female white rabbits (2 kg/10 weeks) purchased from Charles River Laboratories (Wilmington, MA, USA). Rabbits were immunized with 1 mg of MVF chimeric peptide emulsified in Montanide ISA 720 (SEPPIC Paris, France) and 333 μg nor-MDP adjuvant (N-acetyl-glucosamine-3yl-acetyl L-alanyl-D-isoglutamine) and boosted twice at three-week intervals, as previously.Citation54 The vehicle Montanide ISATM 720 was purchased from SEPPIC and it had an approval certificate of analyses for toxicity, emulsifying property, and sterility. N-acetyl-glucosamine-3yl-acetyl-L-alanyl-D-isoglutamine (nor-MDP) was purchased from Penninsula Labs (Torrance, CA).
Female BALB/c mice, 6–8 weeks old, were immunized with 0.1 mg of peptide emulsified in ISA 720 with 33 μg of nor-MDP. Mice were boosted with the respective doses at 3-week intervals. Blood was collected submandibular sera tested for antibody titers. Antibody titers were monitored by indirect ELISA against the peptide immunogens, the peptide B cell epitope, and an acetylated version of the B-cell epitope. Titers are defined as the highest dilution of sera with an absorbance value of 0.2 after subtracting the blank. Sera were collected weekly and animals were euthanized at approximately 18–21 days after tumor challenge. All experiments were performed in accordance with the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals and approved by the Ohio State University Institutional Animals Care and Use Committee and detailed in the accepted protocol.
In vivo studies of four peptide vaccine hPD-1 epitopes: CT26 tumor model in mice
The peptide vaccines were dissolved in sterile water and emulsified in Montanide ISA 720 (1:1) and 33 µg nor-MDP. Female BALB/c mice (Charles River Laboratories, Wilmington, MA) at the age of 6 to 8 weeks were immunized three times at 3-week intervals with 100 µg peptide vaccine, and 2 weeks after the third immunization, the mice were challenged subcutaneously (s.c) with 105 CT 26 tumor cells. PBS treated mice were used as a negative control. Mice were treated twice a week with anti-mPD-1 mAb 29F.1A12 (Bio X Cell, West Lebanon, NH) 200 µg/dose reconstituted in PBS was used as a positive control. Tumor growths were monitored daily and measured twice weekly for up to 21 days after challenge. During immunization, blood was drawn biweekly and used in ELISA to monitor antibody titers.
In vivo studies of peptide vaccine hPD-1 epitopes: CT26/HER-2 tumor model in syngeneic BALB/c mice
Peptide vaccines 100 μg were dissolved in sterile water and emulsified in Montanide ISA 720 (1:1) and 33 μg nor-MDP. Female BALB/c mice (Charles River Laboratories, Wilmington, MA) at the age of 6 to 8 weeks were immunized three times at 3-week intervals with of each peptide vaccine, and 2 weeks after the third immunization, the mice were challenged s.c with CT26/HER-2 tumor cells (105 per mouse). The CT26/HER-2 is a model previously used by us and others for proof-of-principle in active and passive immunotherapy.Citation55–58 Mice were treated twice a week with mPD-1 mAb 29F.1A12 (Bio X Cell, West Lebanon, NH) 200 µg/dose as a positive control and PBS as a negative control. Tumor growths were monitored daily and measured twice weekly for up to 21 days after challenge. During immunization, blood was drawn biweekly and used in ELISA to monitor antibody titers. The mice were euthanized at the end of treatment and tumors and organs (spleen, liver, heart, lung, kidney, and tumor were extracted) Tumors and organs from BALB/c mice vaccinated with combination peptides (HER-2 and PD-1) were collected and submitted for analysis at the Comparative Pathology & Mouse Phenotyping Core facility of the Comprehensive Cancer Center department of Veterinary Biosciences (Pathologist: Krista M. D. La Perle, DVM, PhD, Dipl. ACVP).
ELISA
The 96-well plates (COSTAR, Kennebunk, ME) were coated with 100 µl of antigen at 2 µg/ml in PBS overnight at 4°C. Nonspecific binding sites were blocked for 1 h with 200 µl of PBS-1% BSA, and plates were washed with phosphate-buffered Tween 20 (PBT). Rabbit antiserum (1:500) or mouse antiserum (1:50) in PBT was added to antigen-coated plates in duplicate wells, serially diluted 1:2 in PBT, and incubated for 2 h at room temperature. After washing the plates, 100 µl of 1:500 goat anti-rabbit or goat anti-mouse IgG conjugated to horseradish peroxidase were added to each well and incubated for 1 h. After washing, the bound antibody was detected using 50 µl of 0.15% H2O2 in 24 mM citric acid and 5 mM sodium phosphate buffer (pH 5.2) with 0.5 mg/ml 2,29-aminobis (3-ethylbenzthiazoline-6-sulfonic acid, ABTS) as the chromophore. Color development was allowed to proceed for 10 min, and the reaction was stopped with 25 µl of 1% SDS. Absorbance was determined at 410 nm using a Microplate Reader (BIO-RAD, Hercules, CA). Antibody titers were determined as described previouslyCitation56 and defined as the reciprocal of the highest serum dilution with an absorbance of 0.2 or greater after subtracting background. All data represent the average of duplicate samples.
Mouse isotyping assay
Antibody isotypes (i.e., IgA, IgM, IgG1, IgG2a, IgG2b, and IgG3) were determined using the Mouse Typer Isotyping Kit (BIO-RAD, Hercules, CA). Briefly, wells of a 96-well assay plate were coated with 100 µl of 2 µg/ml peptide antigen in ddH2O and incubated at 4°C overnight. The plate was washed with washing buffer (0.05% tween-20 and 1% horse sera in PBS). The plate was blocked with 1% BSA in PBS at room temperature for 1 h 100 µl of diluted sera was added to each well. After washing the wells, 100 µl ready to use rabbit anti-mouse subclasses antibodies were added to each well, respectively, and incubated at room temperature for 2 h. The wells were washed again, 100 µl (1/3000 dilution of goat anti-rabbit antibody conjugated to Horseradish Peroxidase (HRP) (ABCAM, Cambridge, MA)) was added to each well and incubated for 1 h at room temperature in dark. The plate received a final wash and 50 µl prepared 2,2′-Azino-bis (3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS, Sigma, St Louis, MO) substrate solution was added to each well. The reaction was stopped with 25 µl 1% SDS in ddH2O. Absorbance at 415 nm was determined using an ELISA plate reader. Dilutions of each sera samples were determined by the ELISA titers shown in absorbance of 0.4 or higher after subtracting the background.
Tumor growth and statistical analysis
Tumor sizes were observed daily and measured by calipers. Tumor volumes were calculated by formula: tumor volume = (length x width2)/2 (LWW). Percentage of tumor growth inhibition (TGI%) was defined as the difference between median tumor volume (MTV) of treatment group with the PBS control group and the value was calculated by formula: “TGI% = 100*(MTV control – MTV test)/MTV” control. Complete response (CR) was defined as tumor volume equal or less than 50 mm3 for at least 3 consecutive measurements during the study. The Log-rank (Mantel-Cox) test was used to compare the percentage of CR in multiple groups. The significance of CR was analyzed by Fisher’s exact test. Data statistical analysis was performed by using online tumor growth analysisCitation59 tool app https://kroemerlab.shinyapps.io/TumGrowth/for one-way analysis of variance (one-way ANOVA) to compare multiple groups or difference between two groups. GraphPad Prism 8.1.2 (GraphPad Software, Inc. La Jolla, CA) was used to generate figures and to perform Log-rank (Mantel-Cox) test or two-way ANOVA to analysis whole curve. All values are expressed as means ± standard deviation (SD). p value less than 0.05 was accepted as statistically significantly different. indicate p < .05, indicate p < .01.
Results
Four novel peptide epitopes for hPD-1 are identified, demonstrated high immunogenicity/antigenicity and binding specificity characterized by (SPR)
B-cell epitopes were ranked based on six correlates of antigenicityCitation44 and correlated with their secondary structure, combined analysis of these epitopes with crystal structures complex of human PD-1/human PD-L1 (hPD-1/hPD-L1).Citation34 From this analysis, four B-cell epitope sequences of human PD-1 were identified for further investigation: amino acid 32–50, 45–64, 73–90, and 92–110 (see for peptide sequence). shows the secondary structure of the sequences as modeled using PyMOL 3-D modeling software, and ) shows the structure of the PD-1/PD- L1 complexCitation34. shows the PD-1 (92–110) epitope sequence location in the 3-D structure of PD-1.
Figure 1. Identification of four B-cell epitope sequences of human PD-1. (a) Amino acid sequences of human PD-1, peptides 32–50, 45–64, 73–90 and 92–110 were chosen for evaluation. (b) The secondary structure of the sequences of human PD-1 epitopes as modeled by PyMOL. (c) The structure of the PD-1/PD-L1 complex as adapted by Zak et al.,Citation34 key amino acids involved in the interaction between hPD-1 (light blue ribbon model; navy blue amino acid residues) and hPD-L1 (green ribbon model; light green amino acid residues) are illustrated. Amino acids that constitute the central hydrophobic core of the hPD‐1/hPD-L1 interface are indicated in yellow. Strands on both PD-1 and PD-L1 are indicated by red letters; (d) The 3D structure of human PD-1(92–110) peptide epitope as illustrated by PyMOL. (e): Immunogenicity and antigenicity of MVF-PD-1 B-cell epitopes. New Zealand white rabbits were immunized with 1 mg of each MVF-peptide immunogens dissolved in dd H2O emulsified (1:1) in Montanide ISA 720 vehicle (Seppic) with 333 μg of N-acetylglucosamine-3yl-acetyl-l-alanyl-d-isoglutamine (nor-MDP). Rabbits were boosted with the same doses at 3-week intervals. Blood was collected via the central auricular artery in rabbits. Sera (terminal) from rabbit (3Y+3) immunized with MVF-PD-1 peptide immunogens were tested individually versus the immunogen, acetylated B-cell epitope, the free B-cell epitope and the rhPD-1 protein by ELISA. 200 ng/well peptide or 500 ng/well of rhPD-1protein were used in duplicates to coat the ELISA plates. Titers are defined as the highest dilution of sera with an absorbance value of 0.2 after subtracting the blank
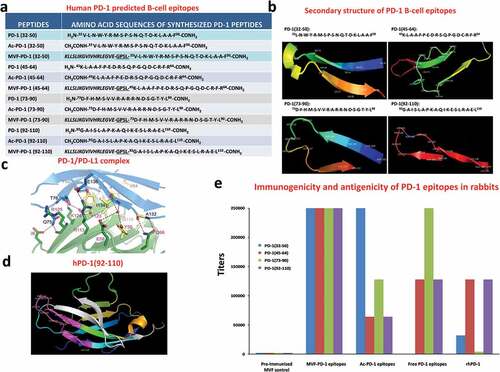
The four peptide sequences were synthesized as chimeric constructs with a “promiscuous” T- helper epitope derived from the measles virus fusion protein (MVF, amino acids 288–302) and their immunogenicity was evaluated in outbred rabbits. All four epitopes elicited high-tittered antibodies >250,000 against the immunizing peptides (MVF-PD-1 peptides, acetylated peptides, and free peptides). More importantly, the results showed that two epitopes (MVF-PD1 (45–64 and 92–110)) elicited high levels of anti-peptide antibodies that recognized the recombinant hPD-1 (rhPD-1) protein (). One epitope 73–90 had little reactive antibodies and another epitope 32–50 had lower antibodies reactive to rhPD-1.
SPR spectroscopy was used to assess the binding specificity that identified three of the four peptide sequences (MVF-PD-1 [45–64; 73–90; and 92–110]) were able to recognize and bind to both recombinant hPD-L1 (rhPD-L1) and nivolumab. Subsequent analyses showed that both the acetylated (Ac) peptides and free peptides bound to both rhPD-L1 and nivolumab (). The remaining peptide sequence, MVF-PD-1(32–50) did not show these properties. Upon rhPD-1 injection, nivolumab generated a strong signal with response (3740 RU), rhPD-L1 caused a weak signal (461 RU), while the human IgG negative control did not show any binding signal (data not shown). BSA did not lead to any binding, indicating PD-1 binding to nivolumab and rhPD-L1 was specific. , panel A1 shows that MVF-PD-1 (45–64), MVF-PD-1 (73–90), MVF-PD-1 (92–110) bind to immobilized hPD-L1, resulting in around 110 RU. In contrast, MVF-PD-1 (32–50) exhibited very weak binding (11 RU), which is similar to the negative control, MVF-HER-2 (266–296) (20 RU). The same three positive MVF-peptides revealed stronger binding to nivolumab, 1030 RU, 1000 RU, 970 RU, respectively (, panel A4). Again the MVF-PD-1 (32–50) showed negligible binding capacity (20 RU) indicating that this sequence does not represent a viable epitope. It was concluded that MVF-PD-1 (45–64, 73–90, and 92–110) were able to recognize both hPD-L1 and nivolumab, while MVF-PD-1 (32–50) does not. Additionally, the acetylated peptides also bind to hPD-L1 and nivolumab, albeit more weakly than the chimeric MVF peptides (, panel A2 and panel A5, respectively). The free peptides also bind both PD-L1 and nivolumab; PD-1 (73–90) shows much stronger binding to rhPD-L1 and nivolumab than PD-1 (45–64) and PD-1 (92–110) (, panel A3 and panel A6, respectively). PD-1 (45–64) is the second strongest binder. Therefore, binding efficiency of free peptides can be ranked as: (73–90) > (45–64) > (92–110). From these binding studies, it can be concluded that the PD-1 peptides 46–64, 73–90, and 92–110 can recognize the rPD-L1 and can act as small peptide inhibitors of PD-1/PD-L1 interaction.
Figure 2. Characterization of novel hPD-1 peptide epitopes by SPR. The specificity of PD-1 was determined by SPR spectroscopy (Biacore T200, at 25°C) and binding affinities to immobilized rhPD-L1 and nivolumab on CM5 sensor chips were determined. rhPD-L1 and nivolumab were immobilized onto the gold surface of a CM5 sensor chip by direct amine coupling. Panels a1-a3 represents the different forms of peptide binding to rhPD-L1 and Panels a4-a6 binding to nivolumab
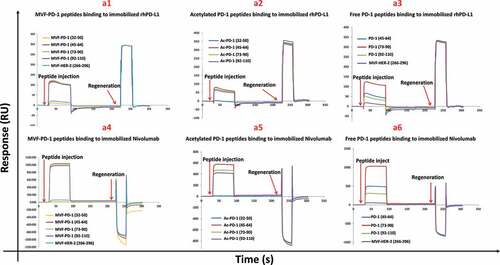
Figure 3. Immunogenicity and preliminary assessment of hPD-1 peptide epitopes in BALB/c mice syngeneic model challenged with CT26 colon carcinoma cell line. (a): Scheme of BALB/c mice vaccination and tumor engraftment. BALB/c mice (5 mice/group) 6–8 weeks old, except PBS group, which has 10 mice, were immunized with MVF-peptide immunogens emulsified in ISA 720 with nor-MDP three times and three weeks apart. Mice were immunized with four MVF-PD-1 vaccine constructs [PD-1(32–50), PD-1(45–64), PD-1(73–90), PD-1(92–110)] prior to tumor challenge. Blood was collected weekly and sera tested for antibody titers by ELISA. Two weeks after the third immunization (3Y), the mice were engrafted with CT26 tumor cells 105 per mouse. Control mice were treated twice weekly with PBS (10 mice as per Figure 4) as negative control and twice weekly with anti-mPD-1 mAb 29F.1A12 as positive control starting 2 days after tumor challenge. Tumor growths were observed twice weekly and measured by calipers; (b) Immunogenicity of MVF-PD-1 peptides in BALB/c mice immunized with various peptide constructs. Sera collected weekly were tittered against each individual MVF-PD-1 peptide immunogen. The terminal bleeds were collected 21 days post challenge prior to mice being euthanized. (c) Individual plots of tumor growths in BALB/c mice immunized with MVF-PD-1 vaccine constructs [PD-1(32–50), PD-1(45–64), PD-1(73–90), PD-1(92–110)], PBS as negative control and anti-mPD-1 mAb (29F.1A12 as positive control. (d) Plots of tumor volume LWW at Day 14 and Day 16 for each of the four treatment groups; Two-way ANOVA was used to analyze the whole curves of tumor growth, which shows significant difference with p < .01; (e) Percentage of tumor growth inhibition (TGI%) was defined as the difference between median tumor volume (MTV) of treatment group with the PBS control group and the value was calculated by formula: TGI% = 100*(MTV control-MTV test)/MTV control which were calculated at Day 14 and Day 16. (f) Complete response (CR) was defined as tumor volume equal or less than 50 mm3 for at least 3 consecutive measurements during the study. The Log-rank (Mantel-Cox) test was use to compare the percentage of CR in multiple groups
![Figure 3. Immunogenicity and preliminary assessment of hPD-1 peptide epitopes in BALB/c mice syngeneic model challenged with CT26 colon carcinoma cell line. (a): Scheme of BALB/c mice vaccination and tumor engraftment. BALB/c mice (5 mice/group) 6–8 weeks old, except PBS group, which has 10 mice, were immunized with MVF-peptide immunogens emulsified in ISA 720 with nor-MDP three times and three weeks apart. Mice were immunized with four MVF-PD-1 vaccine constructs [PD-1(32–50), PD-1(45–64), PD-1(73–90), PD-1(92–110)] prior to tumor challenge. Blood was collected weekly and sera tested for antibody titers by ELISA. Two weeks after the third immunization (3Y), the mice were engrafted with CT26 tumor cells 105 per mouse. Control mice were treated twice weekly with PBS (10 mice as per Figure 4) as negative control and twice weekly with anti-mPD-1 mAb 29F.1A12 as positive control starting 2 days after tumor challenge. Tumor growths were observed twice weekly and measured by calipers; (b) Immunogenicity of MVF-PD-1 peptides in BALB/c mice immunized with various peptide constructs. Sera collected weekly were tittered against each individual MVF-PD-1 peptide immunogen. The terminal bleeds were collected 21 days post challenge prior to mice being euthanized. (c) Individual plots of tumor growths in BALB/c mice immunized with MVF-PD-1 vaccine constructs [PD-1(32–50), PD-1(45–64), PD-1(73–90), PD-1(92–110)], PBS as negative control and anti-mPD-1 mAb (29F.1A12 as positive control. (d) Plots of tumor volume LWW at Day 14 and Day 16 for each of the four treatment groups; Two-way ANOVA was used to analyze the whole curves of tumor growth, which shows significant difference with p < .01; (e) Percentage of tumor growth inhibition (TGI%) was defined as the difference between median tumor volume (MTV) of treatment group with the PBS control group and the value was calculated by formula: TGI% = 100*(MTV control-MTV test)/MTV control which were calculated at Day 14 and Day 16. (f) Complete response (CR) was defined as tumor volume equal or less than 50 mm3 for at least 3 consecutive measurements during the study. The Log-rank (Mantel-Cox) test was use to compare the percentage of CR in multiple groups](/cms/asset/b2019852-2e33-4f7e-9725-12bce924d55a/koni_a_1818437_f0003_oc.jpg)
Figure 4. Efficacy of MVF-PD-1 (92–110) vaccine candidate in BALB/c mice syngeneic model challenged with CT26 colon carcinoma cell line. (a) Scheme of BALB/c mice vaccination and tumor engraftment. BALB/c mice (8-10 mice/group) 6–8 weeks old were immunized with MVF-PD-1(92–110) vaccine emulsified with nor-MDP adjuvant in ISA 720 vehicle. Mice were immunized three times and three weeks apart, 2 weeks after the third immunization (3Y), the mice were engrafted with 105 CT26 tumor cells per mouse. Control mice were treated twice weekly with PBS as negative control (the PBS group shown in this study and that of Figure 3 are the same since both studies were conducted in parallel) and twice weekly with anti-mPD-1 mAb (29F.1A12) as positive control group starting on Day 2 after tumor challenge. Tumor growths were observed twice weekly and measured by calipers; (b) Immunogenicity of MVF-PD-1 (92–110) peptide epitope vaccine in BALB/c mice. Mice bleeds were collected weekly after the primary immunization, and ELISA was used to detect antibody titers in sera. For example the nomenclature is 2Y+1 signifies one week post 2nd injection; 3Y+2 signifies 2 weeks post 3rd injection; (c) Antibody isotypes in BALB/c mice (3Y+1) after immunization with MVF-PD-1 (92–110) nor-MDP and ISA 720; (d) Individual plots of tumor growths by days in BALB/c mice immunized with MVF-PD-1 peptides, PBS was used as negative control and anti-mPD-1 mAb (29F.1A12) was used as positive control; (e) Plots of tumor volume LWW at Day 14 and Day 16 for each of the three treatment groups, which showed statistical differences with p < .01 and p < .05 respectively analyzed by one-way ANOVA; (f) Percentage of complete response (CR) was used to perform Log-rank (Mantel-Cox) test, which showed the p < .01, and only MVF-PD-1(92–110) group observed CR after Day 9 of tumor challenge
Evaluation of immunogenicity of four engineered PD-1 epitopes and anti-tumor activity in syngeneic BALB/c model challenge with CT26 colon carcinoma cell line
We initially evaluated the effects of immunization using the four chimeric peptide vaccine MVF-PD-1 epitopes as inhibitors of tumor growth in vivo using the CT26 tumor model in BALB/c mice. The CT26 tumor model is considered a relevant murine model of colon cancer and has been used in previous studies.Citation60 As shown in , mice received booster immunizations every 3 weeks for a total of three vaccinations. Two weeks after the third vaccination, mice were inoculated with murine colon carcinoma CT26 tumor cells (105) s.c and tumor growth was monitored three times a week. Positive control group on Day 0 were engrafted with 105 CT26 tumor cells s.c and received 200 µg injections twice a week of anti-mouse antibodies directed against PD-1 (anti-mPD-1; mAb 29F.1A12) starting at Day 2 after tumor challenge. We monitored the antibody responses of pooled sera at various intervals 1 week and 3 weeks post each vaccination (2Y+1, 2Y+3, 3Y+1, 3Y+2, 3Y+3) against the vaccine constructs (). All vaccinated mice showed high immunogenicity developing high titers of antibodies to the respective immunogens. Tumor growth of individual mice is shown in . Plots of tumor volume calculated by LWW at Day 14 and Day 16 for each of the four treatment groups are showed, respectively ()). Overall two-way ANOVA was used to analyze the whole curve of tumor growth, which shows significant difference with p < .01. All the peptides immunized groups and mAb treated mice are showed with less tumor burden than PBS group. Even at Day 16, with one-way ANOVA tested all the six groups the p value is slightly beyond 0.05 significant cutoff value, which p < .074. However, the percentage of tumor growth inhibition still ranges from 44% to 48%, except MVF-PD-1(45–64) group only with 28% of TGI at Day 16 ()). Percent of CR stopped at Day 12 vs PBS at Day 9 showed significant difference (). At Day 14, one-way ANOVA gave a p < .01 with six groups comparison. All the individual groups versus PBS showed significant difference (). Comparing peptides with mAb treated group the MVF-PD-1(92–110) showed the best tumor control ability than any other candidate peptide epitopes, with the p< .05 compared with mAb treated group (). Nevertheless, the MVF-PD-1(92–110) peptide vaccine showed the best tumor growth inhibition, which with up to 60% TGI is the highest one of all five groups vs with PBS group (). Overall this suggests that PD-1(92–110) epitope could be a useful inhibitory vaccine.
Efficacy of the PD-1 (92-110) vaccine epitope (PD1-Vaxx) versus anti-mouse PD-1 mAb 29F.1A12 in inhibiting tumor growth in a syngeneic BALB/c model challenge with CT26 colon carcinoma cell line
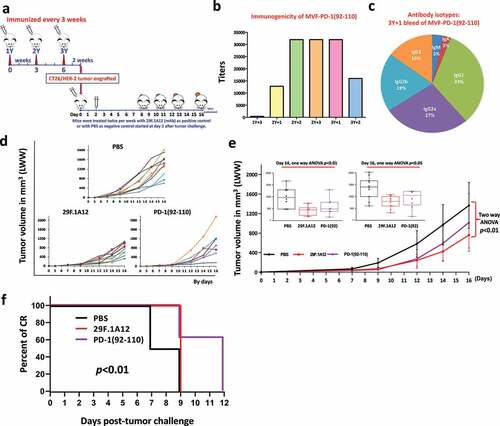
In order to verify the efficacy of the PD1-Vaxx, BALB/c mice (8-10 mice/group) were immunized with MVF-PD-1 (92–110) vaccine emulsified with nor-MDP and ISA 720. Mice were immunized 3 times and three weeks apart ()). Two weeks after the third immunization (3Y), the mice were engrafted with 105 CT26 tumor cells per mouse. Negative control mice were treated with PBS twice weekly and positive control group were treated with mAb 29F.1A12 twice weekly after challenged with 105 CT26 tumor cells. Tumor growth was monitored twice weekly and measured by calipers. The PD1-Vaxx vaccine elicited high titers of PD-1 anti-peptide antibodies ()). We also analyzed the isotypes of antibodies being generated by vaccination () eliciting antibodies predominantly of the IgG1 (33%) isotype followed by IgG2a (27%) and IgG2b (19%). Tumor growth of individual mouse is shown in . The anti-mPD-1 mAb 29F.1A12 and PD1-Vaxx vaccine treatments had similar and significant reduction in tumor growth with two-way ANOVA p < .01 at Day 14 and Day 16 as analyzed by one-way ANOVA as shown in with p< .01 and p< .05, respectively. After Day 9 of tumor challenge, only MVF-PD1(92–110) group observed complete regression. Percentage of CR (Figure 4f) was used to perform Log-rank (Mantel-Cox) test, which showed the p < .01 indicating MVF-PD1(92–110) has a better tumor control ability.
Combination treatment with the PD-1 peptide vaccine (PD1-Vaxx) and combo HER-2 epitopes (B-Vaxx) resulted in robust HER-2 and PD-1 antibody response and effectively inhibits tumor growth in a syngeneic BALB/c CT26/HER-2 model
Next, the transferable CT26/HER-2 tumor modelCitation55 in BALB/c was used to test for synergistic effects of anti-PD-1 immunization therapy in combination with anti-HER-2 immunization therapy to determine whether this combination could increase immunogenicity, enhance anti-tumor responses and provide synergistic benefit in inhibiting tumor growth. Mice were immunized 3 times and three weeks apart ()). BALB/c mice were immunized with single MVF-PD-1 (92–110), combo MVF-HER-2 (266–296) + MVF-HER-2 (597–626) peptides, and triple vaccine combination constructs (MVF-PD-1 (92–110 + MVF-HER-2 (266–296) + MVF-HER-2 (597–626) peptides) emulsified with nor-MDP and ISA 720. Robust HER-2 and PD-1 antibody responses were elicited in all vaccinated mice groups (single, combo, and triple) over the time period to the individual vaccine epitopes as determined by ELISA ()). The isotypes of antibodies elicited in the mice (terminal bleed 21 days post-challenge) were determined to be predominantly of the IgG1 (72% for PD-1; 37% and 75% for HER-2) class followed by IgG2a (18% for PD-1; 23% and 17% for HER-2) ()). Ten days after the final boost CT26/HER-2 tumor cells (105) were transplanted s.c in the flank of each mouse. Negative control mice were treated with PBS while positive control group was treated with anti-mPD-1 mAb (29F.1A12) twice a week after tumor challenge with 105 tumor cells from CT26/HER-2. Tumor growth was monitored and measured with calipers twice a week over the course of the experiments. CT26/HER-2 tumor-bearing mice were euthanized up to 21 days after transplantation. Tumor growth for each mouse is shown in . Tumor sizes at various times are shown in and two-way ANOVA was used to analyze the whole curve of tumor growth, which shows significant difference with p< .01; One-way ANOVA was used to analyze the tumor burden at Day 14 and Day 16, both showing significant difference with p < .01, and all the peptide-immunized groups and mAb-treated group are significantly better than PBS group, with all the p value less than 0.01. Our results show that the triple vaccination (combo HER-2 + PD-1) showed significantly reduced tumor growth in the BALB/c syngeneic model of CT26/HER-2 colon carcinoma versus the single PD-1 vaccine or the combo HER-2 vaccine. More importantly, the triple vaccination showed significant inhibition of tumor growth versus the standard anti-mPD-1 mAb (29F.1A12) positive control (). Significantly higher percentage of tumor growth inhibition (%TGI) was observed in triple combination treatment group, with up to 95% TGI compared to PBS control group treatment at Day 14 and 91% at Day 16 (). Notably, the PD-1 vaccination group showed 78% TGI while mAb 29F.1A12 treatment was 60% TGI and combo HER-2 exhibited 51% TGI. Most importantly, we observed rates of complete response (CR), only in PD-1(92–110) and triple group after Day 14 with 2 out of 10 in PD-1(92–110) treated group versus 9 out of 10 in the triple treated group (), p < .01 was obtained by Fisher’s exact test. In addition, after Day 16, only triple treatment group mice still observed CR mice, which still up to 50% of mice with CR in triple group (). The high rate of CR in the triple treatment group suggests that inhibiting multiple cancer signaling targets with combination of peptide-based vaccines may demonstrate synergistic inhibitory properties as compared to individual treatment options.
Figure 5. Efficacy of combination treatment of MVF-PD-1 (92–110) vaccine and HER-2 combo HER-2 vaccines in syngeneic BALB/c mice challenged with CT26/HER-2 cells. (a) Scheme of mice vaccination and tumor engraftment. 6–8 weeks BALB/c mice were immunized three times three weeks apart with single MVF-PD-1(92–110); combo HER-2: [MVF-HER-2 (266–297) + MVF-HER-2 (597–626)] and triple: (MVF-PD-1 + combo HER-2) peptides emulsified with nor-MDP adjuvant in ISA 720 vehicle. Two weeks after the third immunization (3Y+2), the mice were engrafted with CT26/HER-2 tumor cells 105 per mouse. Control mice were treated twice weekly with PBS as negative control and twice weekly with anti-mPD-1 mAb (29F.1A12) as positive control starting at Day 2 after challenge. Tumor growths were observed twice weekly and measured by calipers; (b) Immunogenicity profile of single, combo and triple vaccinations in BALB/c mice. Mice bleeds were collected weekly after the primary immunization, and ELISA was used to detect antibody titers in sera; (c) Antibody isotypes: The final bleeds (21 days post-challenge at 3Y+2) were used to detect isotypes i.e., IgA, IgM, IgG1, IgG2a, IgG2b and IgG3) in BALB/c mice after immunization with triple constructs: MVF-PD-1 + combo HER-2 were determined using the Mouse Typer Isotyping Kit (BIO-RAD, Hercules, CA); (d) Individual plots of tumor growth in BALB/c mice up to 16 d. (e) Tumor burden by days in BALB/c mice immunized with the different peptide vaccines as illustrated above, PBS was used as negative control and a positive control group with anti-mPD-1 mAb (29F.1A12). Two-way ANOVA (GraphPad Prism 8.1.2) was used to analyze the whole curve of tumor growth, which shows significant difference with p < .01; One-way ANOVA (online analysis toolCitation59 was used to analyze the tumor burden at Day 14 (inset) and Day 16 (inset), respectively; Note: Two-way ANOVA was performed without Day 9 to Day 13 tumor data; (f) Percentage of tumor growth inhibition (TGI%) was used to compare the treatment groups with the control group. The value was calculated by formula: TGI% = 100*(MTV control-MTV test)/MTV control, both calculated at Day 14 and Day 16. (g) Tumor response in mice. The number of mice observed complete CR in each group. Log-rank (Mantel-Cox) test was use to compare the percentage of CR in multiple groups which showed significant difference with p < .01. (h) The CR was only observed in PD-1(92–110) and triple treatment groups after Day 14. We compared the PD-1(92–110) group with triple treatment group by Fisher’s exact test both at Day 14 and Day 16, with p < .01 and p < .05, respectively. Our results show that triple treatment has stronger inhibition of tumor growth n = 9–10 mice in each group
![Figure 5. Efficacy of combination treatment of MVF-PD-1 (92–110) vaccine and HER-2 combo HER-2 vaccines in syngeneic BALB/c mice challenged with CT26/HER-2 cells. (a) Scheme of mice vaccination and tumor engraftment. 6–8 weeks BALB/c mice were immunized three times three weeks apart with single MVF-PD-1(92–110); combo HER-2: [MVF-HER-2 (266–297) + MVF-HER-2 (597–626)] and triple: (MVF-PD-1 + combo HER-2) peptides emulsified with nor-MDP adjuvant in ISA 720 vehicle. Two weeks after the third immunization (3Y+2), the mice were engrafted with CT26/HER-2 tumor cells 105 per mouse. Control mice were treated twice weekly with PBS as negative control and twice weekly with anti-mPD-1 mAb (29F.1A12) as positive control starting at Day 2 after challenge. Tumor growths were observed twice weekly and measured by calipers; (b) Immunogenicity profile of single, combo and triple vaccinations in BALB/c mice. Mice bleeds were collected weekly after the primary immunization, and ELISA was used to detect antibody titers in sera; (c) Antibody isotypes: The final bleeds (21 days post-challenge at 3Y+2) were used to detect isotypes i.e., IgA, IgM, IgG1, IgG2a, IgG2b and IgG3) in BALB/c mice after immunization with triple constructs: MVF-PD-1 + combo HER-2 were determined using the Mouse Typer Isotyping Kit (BIO-RAD, Hercules, CA); (d) Individual plots of tumor growth in BALB/c mice up to 16 d. (e) Tumor burden by days in BALB/c mice immunized with the different peptide vaccines as illustrated above, PBS was used as negative control and a positive control group with anti-mPD-1 mAb (29F.1A12). Two-way ANOVA (GraphPad Prism 8.1.2) was used to analyze the whole curve of tumor growth, which shows significant difference with p < .01; One-way ANOVA (online analysis toolCitation59 was used to analyze the tumor burden at Day 14 (inset) and Day 16 (inset), respectively; Note: Two-way ANOVA was performed without Day 9 to Day 13 tumor data; (f) Percentage of tumor growth inhibition (TGI%) was used to compare the treatment groups with the control group. The value was calculated by formula: TGI% = 100*(MTV control-MTV test)/MTV control, both calculated at Day 14 and Day 16. (g) Tumor response in mice. The number of mice observed complete CR in each group. Log-rank (Mantel-Cox) test was use to compare the percentage of CR in multiple groups which showed significant difference with p < .01. (h) The CR was only observed in PD-1(92–110) and triple treatment groups after Day 14. We compared the PD-1(92–110) group with triple treatment group by Fisher’s exact test both at Day 14 and Day 16, with p < .01 and p < .05, respectively. Our results show that triple treatment has stronger inhibition of tumor growth n = 9–10 mice in each group](/cms/asset/ee4b6d52-e389-4bed-b206-a15bdfb737d2/koni_a_1818437_f0005_oc.jpg)
PD-1 vaccines are safe and do not exhibit toxicity or autoimmunity
All mice vaccinated over a period of 8 weeks showed no signs of scruffiness, lesions, or lethargy. Organs (spleen, liver, heart, lung, kidney, and tumor) from the BALB/c mice vaccinated with combination peptides (HER-2 and PD-1) were collected from mice and submitted for analysis at the Comparative Pathology & Mouse Phenotyping Core facility of the Comprehensive Cancer Center Department of Veterinary Biosciences. No significant lesions were noted in any of the organs submitted for histologic evaluation. There were also no overt biochemical abnormalities noted. All mice had hepatocellular vacuolation consistent with glycogenosis. Glycogen accumulation in the liver is interpreted to be a normal finding and varies depending on the physiological state of the animal. Glycogen accumulation can also be observed as a manifestation of toxicity or with glycogen storage diseases.
Discussion
Currently over a dozen immune checkpoint antibodies targeting CTLA-4 and PD-1/PD-L1 have received FDA regulatory approval worldwide, and there are numerous of ongoing clinical trials for checkpoint inhibitors targeting PD-1 and PD-L1.Citation61–63 These therapies have led to exceptional successes in only a minority of patients. Mechanisms underpinning innate and acquired resistance are responsible in part to for the lack of response to PD-1/PD-L1 blockade, highlighting the rationale that well-designed combination therapies may extend the use and clinical impact. For example, the combination of ipilimumab and nivolumab was approved by the FDA because of positive results for patients with unresectable melanoma and promising Phase I and II trials,Citation64 however the combination also showed increased toxicity compared to monotherapy.Citation65 In this respect, small-molecule inhibitors or peptidomimetics targeting multiple pathways may offer unique advantages such as improved oral bioavailability, prevent immune evasion, and most importantly reduce adverse events.Citation36 This highlights the need to identify new target molecules that could be exploited in the next generation of immunotherapy. Several new antibodiesCitation66,Citation67 targeting other checkpoint LAG-3 and TIM-3, are under clinical investigation either as a monotherapy or in combinations with other immune checkpoint inhibitors or chemotherapy, T-cell vaccines, or radiation therapies.Citation68
In order to address and overcome the inherent limitations and disadvantages of mAbs based immunotherapies, including certain adverse effects and immune-related adverse events, we have developed a portfolio of peptide-based B-cell epitope vaccines focusing on the design of chimeric B- and T-cell novel vaccines to HER-2,Citation54,Citation56,Citation69,Citation70 VEGF,Citation71 EGFR/HER-1,Citation72 HER-3Citation73, and IGF-1RCitation74,Citation75 specifically aimed at eliciting specific high-affinity antibodies (B-cell). We have successfully translated the first generation of HER-2 B-cell peptide vaccines into a Phase I clinical trialCitation76 and have recently successfully completed another Phase I clinical trial (NCI funded and FDA approved Phase I trial IND #14633) with an improved second generation combination HER-2 vaccine trastuzumab-binding epitope (597–626) and the pertuzumab-binding epitope (266–296).Citation37,Citation38 We have thus far completed the dose escalation of our HER-2 combo peptide vaccine, determined the optimal biologic dose, and a Phase II efficacy trial is under way (NCT01376505).
The present manuscript describes the rationale, development, and validation of an alternative checkpoint inhibitor treatment modality that focuses on the innovative discovery of an inhibitory PD-1 B-cell epitope vaccine that elicits a polyclonal antibody response that could mimic nivolumab effects without the concomitant toxicities including immune‐related toxicities and resistance associated with checkpoint blockade antibodies. Successful cancer vaccines require the choice of antigens, an established system designed to elicit a strong effector and memory B- and T-cell responses, and strategies to overcome immune evasion and suppression. The PD-1 vaccine described here showed high immunogenicity and antigenicity to human PD-1 and induced tumor inhibition in vivo in a syngeneic BALB/c colon carcinoma CT26 tumor model. Furthermore, given the great potential for synergistic combinations of immunotherapy with another HER-2 B-cell vaccine. Furthermore, in the CT26/HER-2 model, we demonstrated enhanced immunogenicity and inhibition of tumor growth in the triple combination (combo HER-2 + PD-1) versus the single PD-1 vaccine or the combo HER-2 vaccine. The triple vaccination showed significant inhibition of tumor growth versus the standard anti-mPD-1 mAb (29F.1A12) positive control.Citation43
We also investigated the impact of immunization using a 3 versus 2 weeks schedule as well as the use of additional adjuvant nor-MDP delivered in either ISA 720 or ISA 51. There was little difference between the 2 versus 3-weeks schedule. The forthecoming Phase I trial will be a dose escalating 10 µg, 50 µg, and 100 µg of PD1-Vaxx emulsified in ISA 720. We analyzed the isotypes and cytokines to govern our final composition and immunization protocol for our planned Phase I clinical trial issues. The preponderant isotypes in mice was IgG1 followed by IgG2a. The increase in IgG2a in mice is likely due to T-cell-mediated immune response generated by the “promiscuous” T-cell epitope. Further studies are needed to better understand the role of the different isotypes and T-cell immune response in the mechanism of immune-activation and antitumor activity. Importantly, combination treatment showed no evidence of toxicity or autoimmunity. This suggests that such combination vaccines may have clinical advantages over mAb combination therapy for the treatment of cancer.
To date, concerted efforts have focused on combinations of approved therapies aiming to improve clinical efficacy and further augment positive outcomes and survival. Thus, a variety of combinations of checkpoint inhibitors with other modalities targeting different cell types are being pursued based on the need to block multiple pathways of immunosuppression. Several groups have demonstrated a significant enhancement of antitumor activity by combining PD-1 and CTLA-4 blockade in various tumor models.Citation43,Citation77 The toxicology studies for the ipilimumab/nivolumab combination in cynomolgus monkeys (Macaca fascicularis) revealed additional limited adverse events as compared with individual antibodies.Citation77 However, there are numerous arguments why it is desirable to search for alternatives to mAbs because of the limitations of antibody drugs such as high cost of manufacture, poor oral bioavailability, low tumor permeability, low target selectivity, and intractable adverse events. There is intense research and an urgent need both academically as well as in industry to develop small molecules or low-molecular-weight protein drugs such as peptidomimetics targeting PD-1/PD-L1 blockade to build more efficient and less toxic therapeutics with better pharmacokinetic profiles. The design of these newer molecules is being added by a plethora of published complex crystal structures of PD-1 and PD-L1 inhibitors providing structural information of “hot spots” that laid a solid foundation for drug discovery.Citation78 Compared to PD-1 inhibitors, PD-L1 inhibitors can reduce the incidence of side effects resulting from immune disorders.Citation79 The FDA has approved one humanized mAb (atezolizumab, Tecentric®) and two fully human mAb (avelumab, Bavencio® and durvalumab, Imfinzi®) targeting PD-L1.Citation78,Citation80 The recently reported avelumab/hPD-L1 complex structures have provided clear structural information on how the therapeutic mAbs abrogate the binding of PD-1/PD-L1.Citation81 The PD-L1 targeting antibody, avelumab is now in multiple phase III clinical trials against non-small cell lung cancer (NSCLC) (NCT02395172), advanced renal cell cancer (NCT02684006), and gastric cancer (NCT02625610). A vaccine targeting PD-L1 consisting of the extracellular domain of PD-L1 (PD-L1E) fused to the C-terminal of the translocation domain of diphtheria toxin (DTT) was recently reported.Citation82 The therapeutic efficacy of the DPDL1E vaccine was evaluated in B16-F10 tumor-bearing C57BL/6 mice. The authors concluded that the DPDL1E vaccine targeting PD-L1 induced a PD-L1-specific immune response and delayed tumor growth in vivo. These results indicate a promising avenue for future research in the quest for cancer vaccine design.
Overall, the rational development of various immune strategies for managing certain cancers holds substantial promise for transforming the therapeutic landscape and improving disease management in human beings. Our study also draws attention to the benefits of using a multi-targeted approach, thus it is increasingly appreciated that only use of drugs that affect multiple signaling modalities will result in strong anti-proliferative effects and delay the onset of drug resistance. However, in our current study, we demonstrate that it is possible to generate vaccine combinations that can serve as powerful tools to interrogate oncogenic drivers in cancer cells. To the best of our knowledge there are no B-cell epitope vaccines that target the human PD-1/PD-L1 axis.
The greatest potential significance of our strategy is two-fold: (i) It combines multiple mechanisms of action by activating both B- and T-cell functions and promoting immunological clearance and (ii) It is a targeted approach aimed at inhibiting molecular signaling pathways that are crucial for tumor growth and maintenance. Active immunotherapy offers many advantages, including tumor specificity and the activation of immune responses against antigens that are selectively expressed by tumor cells. Additional benefits of the peptide vaccine approach include the ease and rapid synthesis, safety, lack of toxicity, and cost-effectiveness. Additional advantages of chimeric B- and T-cell vaccines are exquisite specificity and the potential for a durable treatment effect that can be recalled due to immunologic memory. Furthermore, combination anti-PD-1 immunization therapy with anti-HER-2 immunization therapy produced high immunogenicity and greater inhibition of tumor growth. The combined vaccines were safe with no evidence of toxicity or autoimmunity. The development of a PD-1 vaccine in this proposal is highly innovative and when combined with an HER-2 vaccine represents a highly novel promising effective candidate for the treatment of several different metastatic cancers including, colon, breast, and lung.
In conclusion, this engineered MVF-PD-1 (92–110) peptide vaccine (PD1-Vaxx) incorporating a “promiscuous” T-cell epitope derived from measles virus protein MVF epitope represents a vaccine agent useful for clinical translation and highlights the paradigm of B-cell peptide vaccine for future drug development. Toxicity studies for the PD-1 vaccine have been completed successfully in beagle dogs and non-human primates (manuscript in preparation) to support an FDA application for an IND for an upcoming Phase I clinical trial and received two ethical and scientific approvals from the Australian Bellberry Human Research Ethics Committee (HREC) and Macquarie University hospital (NCT04432207) to start Phase 1 trial in Australia. When this trial is completed, we plan to expand the combination of the PD-1 and the combo HER-2 vaccine in a Phase II trial in the future. These results, thus highlight the favorable vaccine strategies of peptide B-cell vaccination for enhanced cancer immunotherapy. The synergistic immunotherapy combinations of HER-2 and PD-1 modalities presented here represent an important opportunity to improve responses and safety outcomes for patients with colon cancer given that HER-2 is also overexpressed in those cancers. The obvious next step would be to apply the combo HER-2 and PD-1 vaccine concept to other HER-2 overexpressing tumors such as breast and GI cancers. We are also examining this combination in BALB/c mice using the mammary tumor line D2F2Citation83 and D2F2/E2 breast cancer cell lines generated by transfection of D2F2 with HER-2.Citation84,Citation85 As we continue to move forward in developing additional B-cell epitope vaccine inhibitors such as PD-L1, LAG-3, and TIM3, significant work remains to be done to determine both the optimal combination regimens as well as predictors of response in order to better personalize treatment. Building upon the progress we have detailed in this manuscript, we believe the field is poised for many more breakthroughs and significant new improvements in the treatment of patients with cancers for maximum clinical benefits.
Author contributions
Conceptualization & Design: Pravin T.P. Kaumaya.
Methodology: Jay Overholser, LinLin Guo, Manuel L. Penichet, Pravin T.P Kaumaya.
Writing: Pravin T.P Kaumaya.
Review and editing: Pravin T.P Kaumaya, Jay Overholser, LinLin Guo, Tanios Bekaii-Saab and Manuel L. Penichet
Disclosure of potential conflicts of interest
Pravin T.P Kaumaya and Tanios Bekaii-Saab are consultants to Imugene, Ltd; All the other authors declare no competing interests.
Acknowledgments
The authors would like to thank Jing Jing Li for performing the Biacore experiments. This study was partly funded by NIH R01CA84356, NIH R21CA13508, and Imugene, Ltd to P.T.P. Kaumaya; NIH R01CA181115 and California Breast Cancer Research Program IDEA Award 25 IB-0053 to MLP.
Additional information
Funding
References
- Ledford H, Else H, Warren M. Cancer immunologists scoop medicine Nobel prize. Nature. 2018;562(7725):20–14. doi:10.1038/d41586-018-06751-0.
- Emens LA, Ascierto PA, Darcy PK, Demaria S, Eggermont AMM, Redmond WL, Seliger B, Marincola FM. Cancer immunotherapy: opportunities and challenges in the rapidly evolving clinical landscape. Eur J Cancer. 2017;81:116–129. doi:10.1016/j.ejca.2017.01.035.
- Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73. doi:10.1186/s12916-016-0623-5.
- Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH, Lao CD. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16(4):375–384. doi:10.1016/S1470-2045(15)70076-8.
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi:10.1056/NEJMoa1504030.
- Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, et al. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. doi:10.1056/NEJMoa1504627.
- Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–1813. doi:10.1056/NEJMoa1510665.
- Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu W-J, Gangadhar TC, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384(9948):1109–1117. doi:10.1016/S0140-6736(14)60958-2.
- Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. doi:10.1056/NEJMoa1507643.
- Yan Y, Kumar AB, Finnes H, Markovic SN, Park S, Dronca RD, Dong H. Combining immune checkpoint inhibitors with conventional cancer therapy. Front Immunol. 2018;9. doi:10.3389/fimmu.2018.01739.
- Kaumaya PT. A paradigm shift: cancer therapy with peptide-based B-cell epitopes and peptide immunotherapeutics targeting multiple solid tumor types: emerging concepts and validation of combination immunotherapy. Hum Vaccin Immunother. 2015;11:1368–1386. doi:10.1080/21645515.2015.1026495.
- Mimura K, Kono K, Hanawa M, Mitsui F, Sugai H, Miyagawa N, Ooi A, Fujii H. Frequencies of HER-2/neu expression and gene amplification in patients with oesophageal squamous cell carcinoma. Br J Cancer. 2005;92:1253–1260. doi:10.1038/sj.bjc.6602499.
- Morrison C, Zanagnolo V, Ramirez N, Cohn DE, Kelbick N, Copeland L, Maxwell LG, Fowler JM. HER-2 is an independent prognostic factor in endometrial cancer: association with outcome in a large cohort of surgically staged patients. J Clin Oncol. 2006;24(15):2376–2385. doi:10.1200/JCO.2005.03.4827.
- Yano T, T, Ohtsu A, Boku N, Hashizume K, Nakanishi M, Ochiai A. Comparison of HER2 gene amplification assessed by fluorescence in situ hybridization and HER2 protein expression assessed by immunohistochemistry in gastric cancer. Oncol Rep. 2006;15:65–71.
- Cirisano FD, Karlan BY. The role of the HER-2/neu oncogene in gynecologic cancers. J Soc Gynecol Investig. 1996;3:99–105.
- Berchuck A, Rodriguez G, Kinney RB, Soper JT, Dodge RK, Clarke-Pearson DL, Bast RC. Overexpression of HER-2/neu in endometrial cancer is associated with advanced stage disease. Am J Obstet Gynecol. 1991;164(1):15–21. doi:10.1016/0002-9378(91)90615-X.
- Kern JA, Schwartz DA, Nordberg JE, Weiner DB, Greene MI, Torney L, Robinson RA. p185neu expression in human lung adenocarcinomas predicts shortened survival. Cancer Res. 1990;50:5184–5187.
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182.
- Rossi E, Grisanti S, Villanacci V, Casa DD, Cengia P, Missale G, Minelli L, Buglione M, Cestari R, Bassotti G. HER-2 overexpression/amplification in Barrett’s oesophagus predicts early transition from dysplasia to adenocarcinoma: a clinicopathologic study. J Cell Mol Med. 2009;13(9b):3826–3833. doi:10.1111/j.1582-4934.2008.00517.x.
- Ross JS, McKenna BJ. The HER-2/neu oncogene in tumors of the gastrointestinal tract. Cancer Invest. 2001;19:554–568. doi:10.1081/CNV-100103852.
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi:10.1056/NEJM200103153441101.
- Marty M, Cognetti F, Maraninchi D, Snyder R, Mauriac L, Tubiana-Hulin M, Chan S, Grimes D, Antón A, Lluch A, et al. Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer administered as first-line treatment: the M77001 study group. J Clin Oncol. 2005;23:4265–4274. doi:10.1200/JCO.2005.04.173.
- Heinemann V, Di Gioia D, Vehling-Kaiser U, Harich H-D, Heinrich B, Welt A, Ziske C, Deutsch G, Pihusch R, Kölbl H, et al. A prospective multicenter phase II study of oral and i.v. vinorelbine plus trastuzumab as first-line therapy in HER2-overexpressing metastatic breast cancer. Ann Oncol. 2011;22(3):603–608. doi:10.1093/annonc/mdq409.
- Ross JS, Slodkowska EA, Symmans WF, Pusztai L, Ravdin PM, Hortobagyi GN. The HER-2 receptor and breast cancer: ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist. 2009;14:320–368. doi:10.1634/theoncologist.2008-0230.
- Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE, Davidson NE, Tan-Chiu E, Martino S, Paik S, Kaufman PA, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353(16):1673–1684. doi:10.1056/NEJMoa052122.
- Smith I, Procter M, Gelber RD, Guillaume S, Feyereislova A, Dowsett M, Goldhirsch A, Untch M, Mariani G, Baselga J, et al. 2-year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: a randomised controlled trial. Lancet. 2007;369(9555):29–36. doi:10.1016/S0140-6736(07)60028-2.
- Gianni L, Dafni U, Gelber RD, Azambuja E, Muehlbauer S, Goldhirsch A, Untch M, Smith I, Baselga J, Jackisch C, et al. Treatment with trastuzumab for 1 year after adjuvant chemotherapy in patients with HER2-positive early breast cancer: a 4-year follow-up of a randomised controlled trial. Lancet Oncol. 2011;12(3):236–244. doi:10.1016/S1470-2045(11)70033-X.
- Slamon D, Eiermann W, Robert N, Pienkowski T, Martin M, Press M, Mackey J, Glaspy J, Chan A, Pawlicki M, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med. 2011;365(14):1273–1283. doi:10.1056/NEJMoa0910383.
- Baselga J, Cortés J, Kim S-B, Im S-A, Hegg R, Im Y-H, Roman L, Pedrini JL, Pienkowski T, Knott A, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366(2):109–119. doi:10.1056/NEJMoa1113216.
- Swain SM, Kim S-B, Cortés J, Ro J, Semiglazov V, Campone M, Ciruelos E, Ferrero J-M, Schneeweiss A, Knott A, et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. The Lancet Oncology. 2013;14(6):461–471. doi:10.1016/S1470-2045(13)70130-X.
- Swain SM, Kim S, Cortes J, Ro J, Semiglazov V, Campone M, Ciruelos E, Ferrero J, Schneeweiss A, Heeson S, et al. Overall Survival (Os) Analysis from the Cleopatra Study of First-Line (1L) Pertuzumab (Ptz), Trastuzumab (T), and Docetaxel (D) in Patients (Pts) with Her2-Positive Metastatic Breast Cancer (Mbc). Ann Oncol. 2014;25:v1. doi:10.1093/annonc/mdu438.7.
- Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3:269–280. doi:10.1038/ncponc0509.
- Yang J, Hu L. Immunomodulators targeting the PD-1/PD-L1 protein-protein interaction: from antibodies to small molecules. Med Res Rev. 2019;39:265–301. doi:10.1002/med.21530.
- Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, Dömling A, Dubin G, Holak TA. Structure of the complex of human programmed death 1, PD-1, and Its Ligand PD-L1. Structure. 2015;23(12):2341–2348. doi:10.1016/j.str.2015.09.010.
- Weinmann H. Cancer immunotherapy: selected targets and small-molecule modulators. ChemMedChem. 2016;11:450–466. doi:10.1002/cmdc.201500566.
- Zhan -M-M, Hu X-Q, Liu -X-X, Ruan B-F, Xu J, Liao C. From monoclonal antibodies to small molecules: the development of inhibitors targeting the PD-1/PD-L1 pathway. Drug Discov Today. 2016;21(6):1027–1036. doi:10.1016/j.drudis.2016.04.011.
- Garrett JT, Rawale S, Allen SD, Phillips G, Forni G, Morris JC, Kaumaya PTP. Novel engineered trastuzumab conformational epitopes demonstrate in vitro and in vivo antitumor properties against HER-2/ neu. J Immunol. 2007;178(11):7120–7131. doi:10.4049/jimmunol.178.11.7120.
- Allen SD, Garrett JT, Rawale SV, Jones AL, Phillips G, Forni G, Morris JC, Oshima RG, Kaumaya PTP. Peptide vaccines of the HER-2/neu dimerization loop are effective in inhibiting mammary tumor growth in vivo. J Immunol. 2007;179:472–482. doi:10.4049/jimmunol.179.1.472.
- Cho H-S, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW, Leahy DJ. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421(6924):756–760. doi:10.1038/nature01392.
- Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, Kofler M, Jorissen RN, Nice EC, Burgess AW, et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol Cell. 2003;11:495–505.
- Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004;5(4):317–328. doi:10.1016/S1535-6108(04)00083-2.
- Bekaii-Saab T, Wesolowski R, Ahn DH, Wu C, Mortazavi A, Lustberg M, Ramaswamy B, Fowler J, Wei L, Overholser J. Phase I immunotherapy trial with two chimeric HER-2 B-cell peptide vaccines emulsified in montanide ISA 720VG and Nor-MDP adjuvant in patients with advanced solid tumors. Clin Cancer Res. 2019;25(12):3495–3507. doi:10.1158/1078-0432.CCR-18-3997.
- Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013;73(12):3591–3603. doi:10.1158/0008-5472.Can-12-4100.
- Kaumaya PTP, Kobs-Conrad S, DiGeorge AM, Stevens V. In:G. M., Basava AnantharamaiahC. editors. PEPTIDES. Boston, US: Springer-Verlag; 1994. p. 133–164.
- Karplus PA, Schulz GE. Refined structure of glutathione reductase at 1.54 A resolution. J Mol Biol. 1987;195:701–729. doi:10.1016/0022-2836(87)90191-4.
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi:10.1016/0022-2836(82)90515-0.
- Hopp TP, Woods KR. Prediction of protein antigenic determinants from amino acid sequences. Proc Natl Acad Sci U S A. 1981;78:3824–3828. doi:10.1073/pnas.78.6.3824.
- Rose GD, Geselowitz AR, Lesser GJ, Lee RH, Zehfus MH. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229:834–838.
- Thornton JM, Edwards MS, Taylor WR, Barlow DJ. Location of ‘continuous’ antigenic determinants in the protruding regions of proteins. Embo J. 1986;5:409–413. doi:10.1002/j.1460-2075.1986.tb04226.x.
- Welling GW, Weijer WJ, van der Zee R, Welling-Wester S. Prediction of sequential antigenic regions in proteins. FEBS Lett. 1985;188:215–218. doi:10.1016/0014-5793(85)80374-4.
- Chou PY, Fasman GD. Prediction of the secondary structure of proteins from their amino acid sequence. Adv Enzymol Relat Areas Mol Biol. 1978;47:45–148.
- Novotny J, Handschumacher M, Haber E, Bruccoleri RE, Carlson WB, Fanning DW, Smith JA, Rose GD. Antigenic determinants in proteins coincide with surface regions accessible to large probes (antibody domains). Proc Natl Acad Sci U S A. 1986;83:226–230. doi:10.1073/pnas.83.2.226.
- Lin DY, Tanaka Y, Iwasaki M, Gittis AG, Su H-P, Mikami B, Okazaki T, Honjo T, Minato N, Garboczi DN. The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc Natl Acad Sci U S A. 2008;105(8):3011–3016. doi:10.1073/pnas.0712278105.
- Dakappagari NK, Douglas DB, Triozzi PL, Stevens VC, Kaumaya PT. Prevention of mammary tumors with a chimeric HER-2 B-cell epitope peptide vaccine. Cancer Res. 2000;60:3782–3789.
- Penichet ML, Challita PM, Shin SU, Sampogna SL, Rosenblatt JD, Morrison SL. In vivo properties of three human HER2/neu-expressing murine cell lines in immunocompetent mice. Lab Anim Sci. 1999;49:179–188.
- Dakappagari NK, Pyles J, Parihar R, Carson WE, Young DC, Kaumaya PTP. A chimeric multi-human epidermal growth factor receptor-2 B cell epitope peptide vaccine mediates superior antitumor responses. J Immunol. 2003;170(8):4242–4253. doi:10.4049/jimmunol.170.8.4242.
- Parihar R, Dierksheide J, Hu Y, Carson WE. IL-12 enhances the natural killer cell cytokine response to Ab-coated tumor cells. J Clin Invest. 2002;110:983–992. doi:10.1172/JCI15950.
- Helguera G, Rodriguez JA, Penichet ML. Cytokines fused to antibodies and their combinations as therapeutic agents against different peritoneal HER2/neu expressing tumors. Mol Cancer Ther. 2006;5:1029–1040. doi:10.1158/1535-7163.MCT-05-0488.
- Enot DP, Vacchelli E, Jacquelot N, Zitvogel L, Kroemer G. TumGrowth: an open-access web tool for the statistical analysis of tumor growth curves. Oncoimmunology. 2018;7:e1462431. doi:10.1080/2162402X.2018.1462431.
- Na HS, Lim YK, Jeong Y-I, Lee HS, Lim YJ, Kang MS, Cho C-S, Lee HC. Combination antitumor effects of micelle-loaded anticancer drugs in a CT-26 murine colorectal carcinoma model. Int J Pharm. 2010;383(1–2):192–200. doi:10.1016/j.ijpharm.2009.08.041.
- Johnson CB, Win SY. Combination therapy with PD-1/PD-L1 blockade: an overview of ongoing clinical trials. Oncoimmunology. 2018;7(4):e1408744. doi:10.1080/2162402x.2017.1408744.
- Balar AV, Weber JS. PD-1 and PD-L1 antibodies in cancer: current status and future directions. Cancer Immunol Immunother. 2017;66(5):551–564. doi:10.1007/s00262-017-1954-6.
- Bylicki O, Paleiron N, Margery J, Guisier F, Vergnenegre A, Robinet G, Auliac JB, Gervais R, Chouaid C. Targeting the PD-1/PD-L1 immune checkpoint in EGFR-mutated or ALK-translocated non-small-cell lung cancer. Target Oncol. 2017;12:563–569. doi:10.1007/s11523-017-0510-9.
- Kyi C, Postow MA. Immune checkpoint inhibitor combinations in solid tumors: opportunities and challenges. Immunotherapy. 2016;8(7):821–837. doi:10.2217/imt-2016-0002.
- Naidoo J, Page DB, Li BT, Connell LC, Schindler K, Lacouture ME, Postow MA, Wolchok JD. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2015;26(12):2375–2391. doi:10.1093/annonc/mdv383.
- Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–2194. doi:10.1084/jem.20100643.
- Woo S-R, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–927. doi:10.1158/0008-5472.CAN-11-1620.
- Seliger B. Combinatorial approaches with checkpoint inhibitors to enhance anti-tumor immunity. Front Immunol. 2019;10:999. doi:10.3389/fimmu.2019.00999.
- Dakappagari NK, Lute KD, Rawale S, Steele JT, Allen SD, Phillips G, Reilly RT, Kaumaya PTP. Conformational HER-2/neu B-cell epitope peptide vaccine designed to incorporate two native disulfide bonds enhances tumor cell binding and antitumor activities. J Biol Chem. 2005;280:54–63. doi:10.1074/jbc.M411020200.
- Dakappagari NK, Sundaram R, Rawale S, Liner A, Galloway DR, Kaumaya PTP. Intracellular delivery of a novel multiepitope peptide vaccine by an amphipathic peptide carrier enhances cytotoxic T-cell responses in HLA-A*201 mice. J Pept Res. 2005;65:189–199. doi:10.1111/j.1399-3011.2005.00212.x.
- Vicari D, Foy KC, Liotta EM, Kaumaya PTP. Engineered conformation-dependent VEGF peptide mimics are effective in inhibiting VEGF signaling pathways. J Biol Chem. 2011;286(15):13612–13625. doi:10.1074/jbc.M110.216812.
- Foy KC, Wygle RM, Miller MJ, Overholser JP, Bekaii-Saab T, Kaumaya PTP. Peptide vaccines and peptidomimetics of EGFR (HER-1) ligand binding domain inhibit cancer cell growth in vitro and in vivo. J Immunol. 2013;191(1):217–227. doi:10.4049/jimmunol.1300231.
- Miller MJ, Foy KC, Overholser JP, Nahta R, Kaumaya PTP. HER-3 peptide vaccines/mimics: combined therapy with IGF-1R, HER-2, and HER-1 peptides induces synergistic antitumor effects against breast and pancreatic cancer cells. Oncoimmunology. 2014;3:e956012. doi:10.4161/21624011.2014.956012.
- Foy KC, Miller MJ, Overholser J, Donnelly SM, Nahta R, Kaumaya PTP. IGF-1R peptide vaccines/mimics inhibit the growth of BxPC3 and JIMT-1 cancer cells and exhibit synergistic antitumor effects with HER-1 and HER-2 peptides. Oncoimmunology. 2014;3:e956005. doi:10.4161/21624011.2014.956005.
- Sanabria-Figueroa E, Donnelly SM, Foy KC, Buss MC, Castellino RC, Paplomata E, Taliaferro-Smith L, Kaumaya PTP, Nahta R. Insulin-like growth factor-1 receptor signaling increases the invasive potential of human epidermal growth factor receptor 2-overexpressing breast cancer cells via Src-focal adhesion kinase and forkhead box protein M1. Mol Pharmacol. 2015;87:150–161. doi:10.1124/mol.114.095380.
- Kaumaya PT, Foy KC, Garrett J, Rawale SV, Vicari D, Thurmond JM, Lamb T, Mani A, Kane Y, Balint CR, et al. Phase I active immunotherapy with combination of two chimeric, human epidermal growth factor receptor 2, B-cell epitopes fused to a promiscuous T-cell epitope in patients with metastatic and/or recurrent solid tumors. J Clin Oncol. 2009;27:5270–5277. doi:10.1200/JCO.2009.22.3883.
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107(9):4275–4280. doi:10.1073/pnas.0915174107.
- Chen T, Li Q, Liu Z, Chen Y, Feng F, Sun H. Peptide-based and small synthetic molecule inhibitors on PD-1/PD-L1 pathway: A new choice for immunotherapy? Eur J Med Chem. 2019;161:378–398. doi:10.1016/j.ejmech.2018.10.044.
- Spagnuolo A, Gridelli C. “Comparison of the toxicity profile of PD-1 versus PD-L1 inhibitors in non-small cell lung cancer”: is there a substantial difference or not? J Thorac Dis. 2018;10:S4065–s4068. doi:10.21037/jtd.2018.09.83.
- Almagro JC, Daniels-Wells TR, Perez-Tapia SM, Penichet ML. Progress and challenges in the design and clinical development of antibodies for cancer therapy. Front Immunol. 2018;8. doi:10.3389/fimmu.2017.01751.
- Liu K, Tan S, Chai Y, Chen D, Song H, Zhang CWH, Shi Y, Liu J, Tan W, Lyu J, et al. Structural basis of anti-PD-L1 monoclonal antibody avelumab for tumor therapy. Cell Res. 2017;27(1):151–153. doi:10.1038/cr.2016.102.
- Lin Z, Zhang Y, Cai H, Zhou F, Gao H, Deng L, Li R. A PD-L1-based cancer vaccine elicits antitumor immunity in a mouse melanoma model. Mol Ther Oncolytics. 2019;14:222–232. doi:10.1016/j.omto.2019.06.002.
- Mahoney KH, Miller BE, Heppner GH. FACS quantitation of leucine aminopeptidase and acid phosphatase on tumor-associated macrophages from metastatic and nonmetastatic mouse mammary tumors. J Leukoc Biol. 1985;38(5):573–585. doi:10.1002/jlb.38.5.573.
- Wei WZ, Shi W-P, Galy A, Lichlyter D, Hernandez S, Groner B, Heilbrun L, Jones RF. Protection against mammary tumor growth by vaccination with full-length, modified human ErbB-2 DNA. Int J Cancer. 1999;81:748–754. doi:10.1002/(sici)1097-0215(19990531)81:5<748::aid-ijc14>3.0.co;2-6.
- Whittington PJ, Radkevich-Brown O, Jacob JB, Jones RF, Weise AM, Wei W-Z. Her-2 DNA versus cell vaccine: immunogenicity and anti-tumor activity. Cancer Immunol Immunother. 2009;58(5):759–767. doi:10.1007/s00262-008-0599-x.