
ABSTRACT
Past failures in clinical trials have dampened the enthusiasm for studying the HGF receptor MET and postponed the development of MET-targeted drugs for cancer therapy. However, new evidence suggests that, at least in liver cancer, MET is still a promising therapeutic target, and may also be a potential target for cancer vaccines. This paper briefly highlights novel research advances in this rapidly-evolving field in the perspective of autophagy, and discusses future directions for further investigation of MET-based cancer therapy and vaccination.
Due to the high mortality rate, liver cancer poses a major threat to human health. However, the pathogenesis of liver cancer has not been fully identified; thus, there is no specific and effective treatment for this disease, resulting in a five-year survival rate of liver cancer patients of less than 5%. Therefore, how to efficiently prevent and control the occurrence and progression of liver cancer has become an important public issue.
As the tissue with the greatest regenerative potential, liver naturally has a few phenotypes very similar to those of tumors, including self-sufficiency in growth signals and limitless replicative potential. The hepatocyte growth factor (HGF) and its receptor MET were identified decades ago and initially found to be closely related to liver regeneration. Subsequent studies confirmed their abnormal expression and sustained activation in a variety of malignant tumors including liver cancer. As an important member of the receptor tyrosine kinase (RTK) family, MET dimerizes upon binding HGF, and then auto-phosphorylates to activate phosphoinositide-3-kinase (PI3K)–protein kinase B (AKT) and other downstream signaling pathways that promote cell proliferation and epithelial-mesenchymal transformation. It has been reported that the HGF-MET signaling pathway regulates multiple aspects of cancer, including its occurrence and development, invasion and metastasis, and therapeutic resistance.Citation1 As a result, there was early development of HGF/MET-targeted drug candidates, such as monoclonal antibodies against the extracellular binding sites of receptors as well as small molecular inhibitors that directly target the intracellular kinase activity center.Citation2,Citation3 Unfortunately, previous attempts to put these drugs into clinical practice failed or were put on hold. Why was targeted therapy against such important targets ineffective? We surmise that the current understanding of the action mechanism of the HGF-MET signaling pathway in liver cancer, especially in mediating drug resistance, is not sufficiently comprehensive.
How should MET be targeted in it-based cancer therapy?
Since the liver is the largest metabolic organ in the body, the occurrence and development of liver cancer, as well as its antagonism to therapeutic stress, inevitably involve reprogramming of energy usage. Energy metabolism in tumors is characterized by the Warburg Effect and glutaminolysis. However, tumor cells may also rely on autophagy, a protective “self-eating” process, to replenish energy and nutrient components. In the event of metabolic dysfunction due to drug attack or restricted nutrient supply, excess intracellular macromolecules or damaged organelles are digested and degraded in auto-lysosomes through the autophagic process and ultimately recycled to maintain the basic requirements for tumor cell survival.
Our recent findings suggest that autophagy plays a critical role in the resistance of liver cancer to HGF/MET-targeted drugs.Citation4 We found that the HGF-MET signaling pathway is crucial for energy metabolism and biosynthesis of liver cancer cells; however, when attacked by conventional HGF/MET-targeted drugs, they can induce autophagy to rebuild a novel biosynthetic pathway that is not dependent on typical metabolism, resulting in drug resistance. Autophagy inhibition can completely block biosynthesis in such treatments, thereby significantly improving the efficacy of HGF/MET-targeted drugs to treat liver cancer. Mechanistically, we found that Y1234/Y1235 of MET, the central sites of its kinase activity, are also involved in the reconstitution of a conserved LC3-binding motif. Although MET enzyme activity can be completely suppressed when attacked by conventional drugs, the concomitant generation of Y1234/Y1235-dephosphorylated MET can induce autophagy by recruiting LC3. In the follow-up study, we confirmed that Y1234/Y1235-nonphosphorylated MET is closely linked to autophagy in clinical tissue samples, and that inhibition of autophagy can significantly enhance the anti-tumor effect of conventional HGF/MET-targeted drugs by blocking biosynthesis. This is potentially a simple, convenient, and practical strategy for improving the effectiveness of cancer therapy while avoiding drug resistance. Furthermore, we successfully prepared high-purity, high-specificity, and high-affinity camelid variable heavy homodimers (VHHs) that target MET, and proposed the novel idea of the VHH pool as one drug candidate.Citation5 We confirmed that, compared to traditional small molecule inhibitors or monoclonal antibody drugs, the anti-MET VHH pool completely blocks the activation and activity of MET kinase and further mediates MET degradation via an endocytic-lysosomal pathway to dramatically decrease its protein level. These effects of anti-MET VHH pool significantly inhibit the formation and growth of various tumors including liver cancer. Moreover, the anti-MET VHH pool avoids antagonism due to MET kinase inhibition-induced MET dephosphorylation-activated autophagy. Hence, in addition to proposing a mechanistic explanation for the failure of HGF/MET-targeted therapies in past clinical trials, we developed the VHH pool as a new approach for treating cancer. These improvements have laid a theoretical and technical foundation for further improving the efficacy of HGF/MET-targeted liver cancer therapies.
Can MET be targeted for cancer prevention?
Although we preliminarily discussed drug resistance in targeted therapy for liver cancer from the perspective of pathway interconversion between tumor metabolism and autophagy, the body’s optimal defense against cancer ought to be spontaneous, persistent, and watchful: This inevitably involves the immune system. In recent years, there has been growing evidence indicating that autophagy is just a biological process to recycle nutritive materials, but that it is closely related to the immune regulation of tumors.Citation6 For instance, autophagy of antigen-presenting cells (APCs) can affect the activation of Toll-like receptors, the presentation of antigens by MHC-II molecules, and immunological synapse formation. Moreover, autophagy in T and B lymphocytes is vital to their differentiation and survival under antigen stimulation. As for tumor cells, autophagy enhances their ability to release immunostimulatory molecules, including but not limited to “find me” signal molecules, like ATP and lysophosphatidylcholine, as well as “eat me” signal molecules, like calreticulin and phosphatidylserine, so that APC progenitor cells are recruited and induced to mature and accelerate antigen uptake. In addition, in ATP, heat shock proteins, and HMGB1-mediated stimulation-dependent manners, autophagy can stimulate APC differentiation in parts of tumors and activate inflammasomes, enhance tumor antigen processing and cross-presentation, as well as increase secretion of interleukin 1beta, thereby regulating the functions of γδ T cells and cytotoxic T cells in the tumor microenvironment (TME). Together, these findings suggest that autophagy can regulate tumor immunogenicity from at least three aspects: the immune system, tumor autoantigens, and tumor-immune interactions in the TME.
This leads to an important question: since MET is so important in regulating autophagy in liver cancer, does MET affect the immunogenicity of liver cancer? After a substantial amount of trial and error, our latest research results show that the interaction between MET and the mechanistic target of rapamycin (MTOR) is the key for suppression of liver cancer immunogenicity. MTOR is a well-known central regulator of autophagy. When environmental nutrients are sufficient, MTOR promotes metabolism and the synthesis of biomacromolecules and inhibits autophagy; in contrast, MTOR activity is inhibited when available nutrients cannot meet the needs of the body, resulting in increased autophagy to keep the body alive.Citation7 More importantly, MTOR not only modulates autophagy by sensing changes in nutrient metabolism, it also participates in the body’s immune response – especially immune regulation in TME.Citation8,Citation9 MTOR regulates the life cycle of dendritic cells, the function of effector T cells, the generation of memory T cells, the differentiation of helper T cells, as well as the activation of natural killer cells, so as to promote the body’s anti-tumor immune response. Inhibition of MTOR can interfere with the aggregation of myeloid-derived suppressor cells, promote the transition from anti-inflammatory tumor-associated macrophages to pro-inflammatory ones, induce the differentiation of regulatory T cells, and make cancer-associated fibroblasts reduce the secretion of cytokines that promote tumor growth and resistance to therapy, thus inhibiting the formation of immunosuppressive TME. Additionally, MTOR inhibition can suppress the expression of programmed cell death 1 ligand 1 (PD-L1), which is up-regulated by activation of the MTOR–AKT signaling pathway in most cancer cells. So far, we have confirmed that MET is a core component of the MTOR complex under physiological conditions. It has been found that in addition to the classical RTK–PI3K–AKT signaling pathway, MET can enter cells via endocytosis and directly binds to the vacuolar ATP synthase (V-ATPase) complex to regulate the activity and function of MTOR. We further revealed that the newly discovered MET–V-ATPase–MTOR signaling pathway, rather than the traditional MET–AKT–MTOR pathway, strongly suppresses the protective efficacy of liver cancer vaccination. Therefore, we identified a PI3K–AKT axis-independent MET–MTOR signaling pathway that determines liver cancer immunogenicity.Citation10
Future perspectives
In past studies, due to limited knowledge, the role of MET was largely attributed to its activity as a receptor kinase. Thus, the widely adopted MET-targeted therapeutic strategy was based solely on inhibition of the activity or activation of MET kinase. It was never considered that, when kinase activity is inhibited or kinase activation is blocked, MET can induce autophagy in a way in which kinase activity is not used, thereby potentially influencing the immune system. Our findings indicate that the role and mechanism of the autophagy-mediated immune response in liver cancer can be systematically analyzed from the perspective of the MET–MTOR signaling pathway. Meanwhile, increased clarity of the relationship between MET-mediated immune escape and the occurrence and development of liver cancer will result in better understanding of the rationale behind tumor immunity. In other words, identification of the precise interplay between the representative features of tumors, like deregulating cellular energetics and avoiding immune destruction, as well as understanding their role in co-promoting tumor growth and survival, from the perspective of MET, can provide an advanced theoretical basis and technical solution for the treatment of tumors.
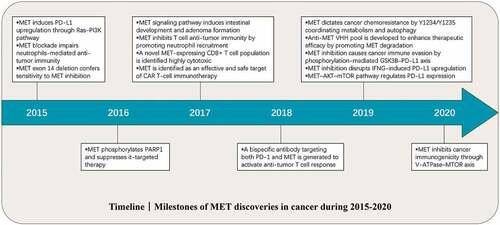
Milestone discoveries related to MET have been made in the last five years (2015–2020) ().Citation1–Citation3 However, considering that both the pathophysiological functions and relevant mechanisms of MET in cancer are not completely clear, further studies including but not limited to the followings are urgently needed: (1) Given that MET plays a compensatory role in acquired resistance of epidermal growth factor receptor (EGFR)-targeted therapy, is EGFR also involved in MET-targeted therapeutic resistance? (2) Are the effects of MET on cancer immunogenicity or other immunity-associated functions autophagy-dependent, even if MET can regulate autophagy? (3) Since a close connection has been found between MET and PD-L1, what is the relationship between MET and other immune checkpoints? (4) What is the translational potential of MET-targeted vaccines in cancer prevention and therapy? (5) What are the clinical prospects of MET-based combination treatment, and how do we maximize the combined benefits to patients?
Figure 1. Timeline: Milestone discoveries relating to MET in cancer research from 2015 to 2020.
Abbreviations
| AKT | = | protein kinase B |
| APC | = | antigen-presenting cell |
| EGFR | = | epidermal growth factor receptor |
| HGF | = | hepatocyte growth factor |
| MTOR | = | mechanistic target of rapamycin |
| PD-L1 | = | programmed cell death 1 ligand 1 |
| PI3K | = | phosphoinositide-3-kinase |
| RTK | = | receptor tyrosine kinase |
| TME | = | tumor microenvironment |
| V-ATPase | = | vacuolar ATP synthase |
| VHH | = | variable heavy homodimer |
Disclosure of potential conflicts of interest
The authors declare no conflicts of interests.
Additional information
Funding
References
- Comoglio PM, Trusolino L, Boccaccio C. Known and novel roles of the MET oncogene in cancer: a coherent approach to targeted therapy. Nat Rev Cancer. 2018;18:341–3.
- Recondo G, Che J, Jänne PA, Awad MM. Targeting MET Dysregulation in Cancer. Cancer Discov. 2020;10(7):922–934. doi:10.1158/2159-8290.CD-19-1446.
- Guo R, Luo J, Chang J, Rekhtman N, Arcila M, Drilon A. MET-dependent solid tumours — molecular diagnosis and targeted therapy. Nat Rev Clin Oncol. 2020;17:569–587.
- Huang X, Gan G, Wang X, Xu T, Xie W. The HGF-MET axis coordinates liver cancer metabolism and autophagy for chemotherapeutic resistance. Autophagy. 2019;15(7):1258–1279. doi:10.1080/15548627.2019.1580105.
- Su Z, Han Y, Sun Q, Wang X, Xu T, Xie W, Huang X. Anti-MET VHH pool overcomes MET-targeted cancer therapeutic resistance. Mol Cancer Ther. 2019;18(1):100–111. doi:10.1158/1535-7163.MCT-18-0351.
- Galluzzi L, Bravo-San Pedro JM, Demaria S, Formenti SC, Kroemer G. Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat Rev Clin Oncol. 2017;14(4):247–258. doi:10.1038/nrclinonc.2016.183.
- Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21:183–203.
- Jones RG, Pearce EJ. MenTORing immunity: mTOR signaling in the development and function of tissue-resident immune cells. Immunity. 2017;46(5):730–742. doi:10.1016/j.immuni.2017.04.028.
- Do MH, Wang X, Zhang X, Chou C, Nixon BG, Capistrano KJ, Peng M, Efeyan A, Sabatini DM, Li MO. Nutrient mTORC1 signaling underpins regulatory T cell control of immune tolerance. J Exp Med. 2020;217(1):e20190848.
- Huang X, Xu X, Wang X, Tang T, Li E, Zhang X, Xu J, Shen H, Guo C, Xu T, et al. The AKT-independent MET-V-ATPase-MTOR axis suppresses liver cancer vaccination. Signal Transduction Targeted Ther. 2020;5(1):122. doi:10.1038/s41392-020-0179-x.