
ABSTRACT
Chimeric antigen receptor T (CAR-T) cell therapy has been applied successfully in treating hematologic malignancies; however, it shows very limited efficacy in treating solid tumors. Adenosine is one of the key immunosuppressive metabolites in tumor microenvironment (TME) of solid tumors. Although the effect of adenosine has been well studied using mouse CAR-T cells, its effect on human CAR-T cells has not been fully elucidated. In particular, there was no evaluation of the CAR-T cells with blocked adenosine signaling in tumor xenograft animal model, which is essential for determining the feasibility of future clinical trials. In this study, we found the expression of A2a receptor (A2AR) and A2b receptor (A2BR) both upregulated in human-derived CAR-T cells, and only A2AR was responsible for adenosine-induced impairment of CAR-T cell function. Disrupting A2AR gene in human CAR-T cells with CRISPR-Cas9 increased the anti-tumor function and prevented the exhaustion of CAR-T cells in vitro. Furthermore, CRL5826-CDX model and two patient-derived xenograft solid tumor models were applied to evaluate the efficacy of A2AR knock-out CAR-T cells, which showed superior capability of inhibiting tumor growth. Taken together, these results demonstrate that A2AR knock-out CAR-T cells have the potential of being an improved CAR-T cell therapy in treating solid tumors.
KEYWORDS:
Introduction
Chimeric antigen receptors T (CAR-T) cell therapy is a promising treatment for cancer.Citation1–4 The CAR is a synthetic transmembrane protein that couples a single-chain variable fragment (scFv) ecto domain to intracellular T-cell signaling domains, thereby redirecting T lymphocytes to cells expressing this antigen. While CAR-T cells showed great efficacy in treating hematological malignancies, Citation3–6 there were very few clinical studies reporting the success in treating solid tumors.Citation7–9 The immunosuppressive nature of tumor microenvironment (TME) is considered to be one of the key factors limiting CAR-T cell efficacy in solid tumor treatment.Citation10–12
Within TME, the immunosuppressive metabolite adenosine is produced by tumor and Treg cells at a relatively high level. Adenosine is the product of a stepwise hydrolysis of adenosine triphosphate through the extracellular nucleotide hydrolases CD39 and CD73, the expression of which is often negatively correlated with the prognosis of patients.Citation13 Adenosine binds to adenosine receptors, transmembrane G protein-coupled receptors including A1, A2A, A2B, and A3, Citation14 to exert immunosuppressive effects. T cells, including CTL, mainly express A2AR and A2BR. After binding to the receptors, adenosine reduces the expression of key proteins such as interferon-γ (IFN-γ) and granzyme by inhibiting the Ras-MAPK kinase-AP1 signaling pathway, thus inhibiting the tumor cell killing ability of CTL.Citation15,Citation16 In addition, inhibiting adenosine signaling with A2AR small molecular inhibitorsCitation17–19 or A2AR-targeted shRNACitation19,Citation20 has all been shown to facilitate the antitumor function of T cells.
In this study, we characterized the suppressive effects of adenosine on human CAR-T cells, and confirmed that CAR-T cells mainly respond to high adenosine in TME through A2AR. We eliminated the negative effects of adenosine on CAR-T cells by knocking out A2AR gene using CRISPR-Cas9 and enhanced the anti-tumor efficacy of CAR-T cells both in vitro and in CDX model and patient tumor-derived xenograft (PDX) models. Our data demonstrate that blocking adenosine signaling by gene editing is a promising strategy to improve the efficacy of CAR-T cells in treating solid tumors.
Results
Adenosine suppressed the cytolysis ability and cytokine production of CAR-T cells
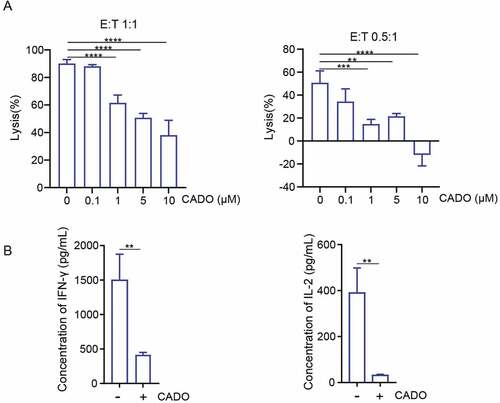
To generate CAR-T cells recognizing mesothelin antigens, we constructed a CAR composed of a fully human scFv (P4) recognizing mesothelin along with CD28 and CD3ζ signaling domain (P4 CAR).Citation21,Citation22 To confirm the specificity of P4 CAR-T cells, we examined the ability of P4 CAR-T cells to lyse CRL5826 (human lung cancer expressing mesothelin) [Fig. S1A] and SKBR3 (human breast cancer without mesothelin expression) cells [Fig. S1B]. We found P4 CAR-T cells recognized and killed mesothelin+ tumor cells [Fig. S1C–D]. P4 CAR-T cells were cocultured with CRL5826 in the presence or absence of various doses of 2-chloroadenosine (CADO), a stable adenosine analog, under two different effector to target (E:T) ratios. Tumor cell killing by P4 CAR-T cells was inhibited in the presence of CADO in a dose-dependent manner [)], and the cytokine IFN-γ and interleukin-2 (IL-2) secretion of P4 CAR-T cells were reduced in the presence of CADO as well [)]. These results confirmed that CADO could inhibit the tumor cell killing capacity and the cytokine release of CAR-T cells.
Figure 1. Adenosine limits the cytolysis ability and cytokine production of CAR-T cells. (a) Specific lysis of P4 cells after incubation with CRL5826 at 1:1 E:T ratio and 0.5:1 E:T ratio with 3 d in the presence of 0, 0.1, 1, 5, and 10 µM CADO. (b) Cytokine IFN-γ and IL-2 production by P4 cells cocultured 3 d with CRL5826 at 0.5:1 E/T ratio in the presence of 0 and 5 µM CADO. **P < .01; ***P < .001; ****P < .0001 were determined by one-way ANOVA test in (a) and unpaired Student’s t-test in (b). Data were represented as mean ± s.d. of three technical replications per assay. The assays were repeated three times.
A2AR is the main receptor responsible for the adenosine-induced CAR-T cell function inhibition
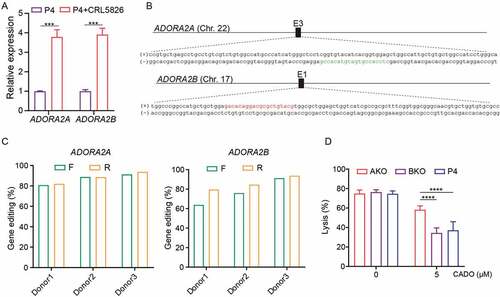
Among the four adenosine receptors, A2A adenosine receptor (A2AR) and A2B adenosine receptor (A2BR) are predominantly expressed in T cells. To evaluate their function, we quantified the A2AR and A2BR expression of P4 CAR-T cells cultured alone or cocultured with CRL5826 cells. Upon encountering tumor antigen, the expression of both A2AR and A2BR was upregulated in P4 CAR-T cells [)]. To test which receptor is responsible for the adenosine-induced impairment of CAR-T function, we generated A2AR knock-out (AKO) and A2BR knock-out (BKO) P4 CAR-T cells using CRISPR-Cas9, with knock-out efficiencies about 75–90% [ and S2A]. The top five off-target sites for each sgRNA were predicted using CRISPOR, Citation23 and we did not detect mutation at any of these loci using TIDE analysisCitation24 (Table S1). The basic characteristics of AKO, BKO, and P4 CAR-T cells were similar with regard to their proliferation ability, ratio of CD4/CD8, and the transduction efficiency of CAR [Figure S2B–D]. AKO, BKO, and P4 CAR-T cells had similar cytolysis ability after being cocultured with CRL5826 in the absence of CADO [)]. In the presence of CADO, the cytolysis of P4 and BKO cells was significantly reduced, while AKO cells had notably higher tumor cell killing capability [)]. These data indicated that the engagement of A2a receptor by adenosine resulted in the impairment of the anti-tumor function of CAR-T cells.
Figure 2. Adenosine-A2AR signaling pathway accounts for the CAR-T cells inhibition. (a) Expression changes of A2AR and A2BR genes in P4 cells under normal culture condition or after cocultured with CRL5826 at 2:1 E:T ratio with 1 d. (b) Schematic diagram of A2AR and A2BR sgRNA in genome. The red indicates the sgRNA targeting sites on the sense strand, and the green color represents antisense strand. (c) Amount of A2AR and A2BR gene disruption measured by TIDE assay on DNA amplified from AKO, BKO, and P4 cells. F and R represent two different direction of Sanger sequencing used for TIDE. (d) Specific lysis of AKO, BKO, and P4 cells after incubation with CRL5826 at 0.5:1 E:T ratio 3 d in the presence of 0 and 5 µM CADO. Comparisons were made between the three groups. ***P < .001; ****P < .0001 were determined by two-way ANOVA test. Data were represented as mean ± s.d. of three technical replications per assay. The assays were repeated two times in (a) and three times in (d).
A2AR knock-out enhanced the anti-tumor function of CAR-T cells in vitro
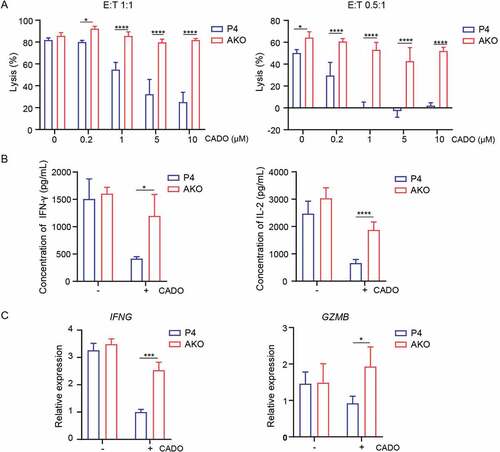
To further characterize the adenosine-A2AR mediated inhibition of CAR-T cell function, we used both CADO and CGS21680 (short for CGS, a specific agonist of A2AR), to model adenosine immunosuppressive microenvironment. AKO and P4 CAR-T cells were cocultured with CRL5826 in the presence or absence of various doses of CADO and CGS. Consistent with the results of using CADO [) and )], tumor cell killing by P4 CAR-T cells was inhibited induced by CGS in a dose-dependent manner [Fig. S3A]. Compared to wild-type P4, A2AR knock-out completely rescued the negative effect of CADO and CGS on the tumor lysis capability [) and S3A]. Meanwhile, the cytokine IFN-γ and IL-2 secretion of AKO cells were improved compared to P4 CAR-T cells in the presence of CADO and CGS [) and S3B]. Consistently, cytolysis-related gene expression of IFNG and GZMB was markedly decreased in P4 CAR-T cells after CADO and CGS treatment () and S3C), while the expression of both genes was increased in A2AR knock-out cells [) and S3C]. These results showed that A2AR knock-out rescued the adenosine-mediated inhibition of CAR-T anti-tumor function.
Figure 3. A2AR knock-out enhanced the anti-tumor function of CAR-T cells in vitro. (a) Specific lysis of AKO and P4 cells after incubation with CRL5826 at 1:1 E:T ratio and 0.5:1 E:T ratio with 3 d in the presence of 0, 0.2, 1, 5, and 10 µM CADO. (b) Cytokine IFN-γ and IL-2 production by AKO and P4 cells cocultured 3 d with CRL5826 at 0.5:1 E/T ratio in the presence of 0 and 5 µM CADO. (c) Expression changes of IFNG and GZMB in AKO and P4 cells after incubation with CRL5826 at 0.5:1 E:T ratio with 3 d in the presence of 0 and 5 µM CADO confirmed by qPCR. *P < .05; ***P < .001; ****P < .0001 were determined by two-way ANOVA test. Data were represented as mean ± s.d. of three technical replications per assay. The assays were repeated three times in (a) and two times in (b–c).
Given that A2AR knock-out significantly increased the efficacy of CAR-T cells, we used SKBR3 to assess the safety of A2AR knock-out P4 CAR-T cells. Similar to P4 CAR-T cells, AKO cells did not kill SKBR3 cells either in the presence or absence of CADO, suggesting the lack of nonspecific toxicity [Fig. S3D].
A2AR knock-out P4 CAR-T cells retain anti-tumor function upon repetitive tumor challenges in the presence of adenosine
Upon infiltrating solid tumors, T cells are likely activated repetitively by tumor cells. This may lead to T-cell exhaustion, which is manifested by impairment of T-cell proliferation and effector function, and the increased immune checkpoint genes expression.Citation25,Citation26 To better model T-cell exhaustion, we used an in vitro tumor re-challenge assay, in which CAR-T cells were harvested and recursively transferred to culture dishes seeded with tumor cells every 48 h, maintaining a constant 2:1 E:T ratio with or without the addition of CADO and CGS [)]. Under multiple rounds of antigen challenges, the proliferation of CAR-T cells was inhibited in all conditions, while the addition of CADO and CGS worsened this inhibition, particularly at round 2 [) and S4A]. Knocking out A2AR made CAR-T cells resistant to the inhibition of CADO and CGS [) and S4A].
Figure 4. A2AR knock-out released CAR-T cells from exhaustion induced by adenosine. (a) Scheme of repetitive tumor challenge assay. (b) Proliferation of AKO and P4 cells after repetitive tumor challenge in the presence of CADO. (c) Specific lysis by AKO and P4 cells after three rounds of CRL5826 challenge in the presence of CADO. (d) Immune checkpoint gene expression of AKO and P4 cells after three rounds of CRL5826 stimulation in the presence of CADO. *P < .05; **P < .01; ****P < .0001 were determined by two-way ANOVA test. Data were represented as mean ± s.d. of three technical replications per assay. The assays were repeated three times in (b–c) and two times in (d).
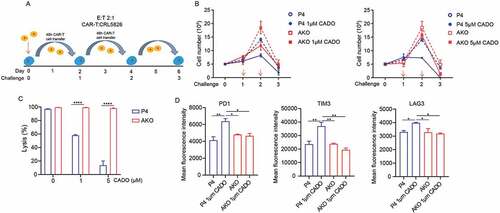
The tumor lysis ability of CAR-T cells was decreased after three rounds of challenges in the presence of CADO and CGS, while AKO cells maintained their activity [) and S4B]. In addition, CADO and CGS upregulated the expression of immune checkpoint genes PDCD1, TIM3, and LAG3 in CAR-T cells after three rounds of challenges, while knock-out of A2AR reduced this effect [) and S4C]. These results indicated that knocking out A2AR made CAR-T cells less prone to T cell exhaustion.
A2AR knock-out CAR-T cells showed superior tumor-eliminating capability in vivo
Next, we tested the anti-tumor efficacy of A2AR knock-out P4 CAR-T cells in CRL5826-CDX model. CD39 and CD73 were key enzymes in adenosine production, with CD73 being the common factor of all major cellular adenosine-producing systems.Citation27,Citation28 We confirmed the expression of CD73 in CRL5826 cells [Fig. S5A]. When the tumor volume reached 300–400 mm3, we infused 5 × 106 CAR-T cells twice with 1 week interval [)]. Compared to the P4 and phosphate buffered saline (PBS) groups, AKO injection limited the tumor growth more efficiently [Fig. S5B]. The tumors were notably smaller in AKO group than in P4 group at d 50, and the weight of the tumors derived from AKO group was lower than P4 group [Fig. S5C]. The engraftment and persistence of CD3+ T cells in the P4 and AKO-treated CDX models had no significant difference [Fig. S5D].
Figure 5. A2AR knock-out P4 CAR-T enhanced tumor killing in PDX models in vivo. (a) Scheme of in vivo assay to test the anti-tumor function of AKO cells. (b) Mesothelin and CD73 expression in two PDX models. (c) Fold changes of tumor volume after CAR-T cells intratumor injection in #1 PDX model. The translucent line represented the tumor volume fold for individual mice, the solid line indicated the mean value. (d) The size and weight of resected tumors derived from mice sacrificed at 70 d after CAR-T cells injection in #1 PDX model. (e) Analysis of T cell types in peripheral blood at 42 d after CAR-T cell injection in #1 PDX model. Comparison was made with CD3+ cells or CD8+ cells in P4 and AKO group. (f) Fold changes of tumor volume after CAR-T cells intratumor injection in #2 PDX model. The translucent line represented the tumor volume fold for individual mice, and the solid line indicated the mean value. **P < .01 was determined by two-way ANOVA test. Data were represented as mean ± s.d.
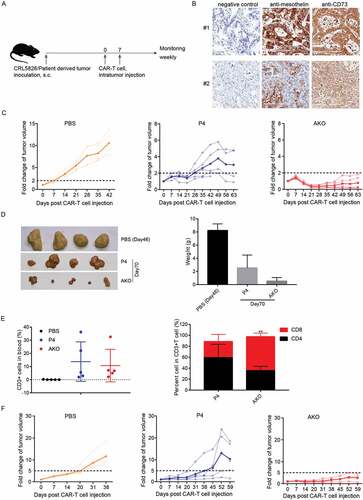
To assess CAR-T cell efficacy in models more relevant to primary tumor, we used two pancreatic-cancer-patient-derived tumor xenograft (PDX) models, which closely retain the structural, morphological, and molecular features of the primary tumor. In PDX sample #1, mesothelin and CD73 were highly and uniformly expressed [)].
Compared to the fast increase of tumor size in PBS group, P4 CAR-T cell injection slowed down the tumor growth. However, at d 63 after injection, the tumor still grew to about three times of the original volume on average. In contrast, 21 d after the first injection, AKO injection reduced the tumor to less than 30% of initial volume, maintaining a relatively low tumor burden afterward [)]. Notably, smaller tumors were observed in AKO group than in P4 group at d 70 when tumors were resected, and the weight of the tumors derived from AKO group was lower than P4 group [)]. The percentages of P4 and AKO CAR-T cells in peripheral blood were similar, while the ratio of CD8 versus CD4 was notably higher in AKO compared to P4 cells [)].
Considering that tumor antigens were not uniformly expressed in many tumors, we evaluated the performance of CAR-T cells in PDX model #2, in which the expression of mesothelin was more heterogenous [)]. The same results were obtained with AKO cells showing apparent anti-tumor advantages by controlling the tumor growth compared to P4 group [)]. There was no difference in the proportion of CD3+ T cells between the P4 and AKO groups in PDX model #2 [Fig. S5E]. In summary, these findings indicated that A2AR knock-out CAR-T cells could produce more potent antitumor activity in vivo.
Discussion
CAR-T cell therapy has produced great clinical results in treating hematopoietic malignancy, Citation3–Citation6 while it has limited efficacy against solid tumors. Immunosuppressive TME of solid tumors is one of the main causes. Attempts to overcome this limitation, including expressing heparinase to degrade the extracellular matrix (ECM), Citation29 knocking out transforming growth factor-β (TGFβ) receptors,Citation22 or expressing dominant-negative TGFβ receptorsCitation30 to resist TGFβ-rich environment, using CAR-Target fibroblast activation protein (FAP) antigen to deplete fibroblast cells,Citation31 or engineering CAR-T cells lacking immune checkpoint genes, Citation32–35 have shown promise in animal models.
As a soluble metabolite, adenosine plays an important role in establishing the immunosuppressive TME, which is conducive to the development of tumors.Citation28 The hypoxia in the tumor tissue facilitates adenosine accumulation in TME. On one hand, hypoxic environment increases the expression of CD39 and CD73, key genes in adenosine production pathway.Citation27,Citation28 On the other hand, hypoxia can reduce the expression of adenosine kinase and inhibit the degradation of adenosine.Citation36,Citation37
We found adenosine receptors A2A and A2B were upregulated in CAR-T cells stimulated by mesothelin positive cells, which was consistent with previous studies.Citation15,Citation16 By knocking out each receptor using CRISPR-Cas9, we found that adenosine exerted immunosuppressive effects mainly through A2AR in human CAR-T cells, with A2BR being less important (). This finding is consistent with previous literature showing that A2AR has an important effect in regulating T cell immunity, Citation15,Citation19,Citation28 while A2BR might be associated with dendritic cells maturation and then influence T cell immunity.Citation38
Previous studies demonstrated that genetic deletion of A2ar in mice or treatment with A2AR antagonist improved the efficacy of adoptive cell transfer therapy.Citation18,Citation19 Mouse-derived CAR-T cells with either genetic, pharmacological, or shRNA targeting of A2AR were proved to improve CAR-T cell efficiency.Citation20 However, the in vitro and in vivo function of A2AR in human-derived CAR-T cells have not been studied in great detail. To provide key information for evaluating the feasibility of clinical studies, we evaluated the effects of adenosine-A2AR pathway on human CAR-T cells, and showed that the presence of adenosine analogs during coculture of human CAR-T cells with tumor cells resulted in a decreased tumor cell killing, reduced expression of IFNG, GZMB, and reduced release of cytokines IFN-γ and IL-2, which was similar to the findings in mouse CAR-T cells. Different from the knockdown gene expression by shRNA, we utilized gene-editing tool to obtain A2AR knock-out human-derived CAR-T cells. Complete blocking of adenosine effects by knocking out A2AR using CRISPR-Cas9 improved the antitumor activity of CAR-T cells in vitro. Furthermore, A2AR knock-out CAR-T cells showed better resistance to exhaustion upon repetitive antigen challenges. Importantly, our data showed that A2AR knock-out human CAR-T cells had an improved efficacy in treating human primary xenograft in PDX models. These encouraging preclinical data showed the potential to move this gene editing improved CAR-T cells into future clinical studies.
The TME of solid tumor is complex; the heterogeneity of antigen and the pathological structure of tumor may affect the therapeutic effect of CAR-T cells. In order to better evaluate the function of A2AR knock-out CAR-T cells, we established two PDX models using different pancreatic cancer specimens. The antigen expression is strong and uniform in one sample, while weaker and heterogenous in the other. But regardless of this difference, A2AR knock-out CAR-T cells effectively inhibited tumor growth and maintained a low tumor load. Although having better efficacy than P4 cells, AKO CAR-T cells were not able to completely clear the tumors. In comparison, our previous study showed that TGFβRⅡ-knock-out CAR-T cells can completely clear the tumor in these two PDX models.Citation22This suggested that multiple immunosuppressive mechanisms coexist within the TME, some of which may play more dominant roles.
In our study, during multi-round antigen challenge assay, in the presence of CADO and CGS, CAR-T cells had an increased PD1 expression, with reduced tumor killing and proliferation, residing at a hypofunctional state similar to the state of T cell exhaustion. The PD1 expression of CAR-T cells remained at a lower level after knocking out A2AR, suggesting that adenosine-A2AR and PD1 pathway may interact together. And, previous study also demonstrated that the antitumor effect was significantly enhanced when anti-PD1 and A2AR inhibitors were used together in mouse model.Citation20 Therefore, blocking adenosine signaling could be combined with other interventions to achieve more potent therapeutic effects.
Blocking the adenosine-A2AR signaling using gene editing renders CAR-T cells resistant to adenosine-rich TME, while avoids systemic toxicity caused by the administration of small molecule A2AR inhibitors. The inhibition of specifically combined pathways in CAR-T cells will likely lead to better efficacy in treating a particular type of TME. As multiplex gene editing using CRISPR-Cas9 has been well established,Citation32,Citation33 blocking multiple inhibitory pathways in CAR-T cells by knocking out several genes simultaneously is a promising strategy for further development. The combination of CAR-T and CRISPR-Cas9 will provide a more targeted, personalized and potent tumor immunotherapy.
Materials and methods
Primary human T cell isolation and CAR-T cell production
Fresh umbilical cord blood units were obtained from Beijing Cord Blood Bank (Beijing, China) from healthy volunteer donors with informed consent. Mononuclear cells were isolated by human mononuclear cells separation medium 1.007 (Beijing Dong Fang Hua Hui Biomedical Technology Co., Ltd., Beijing, China) gradient separation. T cells were isolated using the EasySep human T cell enrichment kit (Stemcell Technologies, Vancouver, Canada). T cells were activated with anti-CD3/CD28 Dynabeads (Thermo Fisher Scientific, Waltham, Massachusetts, USA) at a bead to cell ratio of 1:1 and cultured in X-VIVO15 medium (Lonza, Basel, Switzerland) supplemented with 5% (v/v) heat-inactivated fetal bovine serum (GIBCO) and 300 IU/mL human recombinant IL-2 (Sino Biological Inc., Beijing, China). After 24 h activation, T cells were transduced with lentiviral supernatants, harboring anti-mesothelin CAR. The transfection efficiency was determined 2 d later by flow cytometric analysis. Cell culture medium was replaced by fresh complete medium every 2 d.
Generation of A2AR and A2BR knock-out CAR-T cells
sgRNAs were designed by CRISPOR (http://crispor.tefor.net/). Nucleotide sequence including targeting sequences and T7 promoter were synthesized as forward primer. pX330 plasmid (Addgene #4223, Watertown, Massachusetts, USA) was used as template. The PCR product was used as the template to conduct RNA in vitro transcription using MEGAshortscript T7 kit (Thermo Fisher Scientific). sgRNAs were purified with MEGAclear columns (Thermo Fisher Scientific) and then eluted in RNase-free water. Two days after transfection, after beads were removed by magnetic separation, 1 × 106 CAR-T cells were electroporated with RNP containing Cas9 protein and sgRNA using P3 Primary Cell 4D-Nucleofector X Kit (Lonza), program EO-115, and Nucleofector System N (Lonza). After electroporation, cells were resuspended in pre-warmed complete medium and maintained at 37°C in a humidified atmosphere containing 5% CO2.
Analysis of gene editing efficiency
The indel frequencies of A2AR and A2BR were measured by TIDE (tracking of indels by decomposition) analysis and clonal sequence analysis. The genomic DNA was extracted from edited cells. PCR products were ligated to pEASY vector (TransGen Biotech, Beijing, China), and then transformed in Escherichia coli. Single clone was picked and sequenced to detect mutants. The primers used for the amplification of target loci were listed in Table 2. The levels of genomic disruption of A2AR and A2BR were also confirmed by Sanger sequencing and sequencing results were analyzed by TIDE method (https://tide.deskgen.com/).Citation24
The top five off-target sites for each sgRNA were predicted by CRISPOR (http://crispor.tefor.net/).Citation23 The off-target efficiencies were analyzed by TIDE. Primers used for off-target measurement were listed in Table S2.
Luciferase-based cytolysis assay
The cytotoxicity of the CAR-T cell was assessed by the luciferase-based cytotoxicity assay as described previously.Citation22 Briefly, CRL5826-luciferase cells were suspended at 1 × 105 cells/mL in RPMI1640 medium and cocultured with CAR-T cells at indicated effector to target ratios, then seeded in white luminometer plate followed by maintaining at 37°C in a humidified atmosphere containing 5% CO2. A volume of 10 μL substrate (Promega, Madison, Wisconsin, USA) was added at the indicated incubation time, and then the luminescence was determined by PerkinElmer VICTOR X3 (Walsham, Massachusetts, USA). The results were reported as the percentage of killing based on the luciferase activity of remaining tumor cells (% killing = 100−((RLU from well with effector and target cell coculture)/(RLU from well with target cells) × 100)).
RT-PCR
Cells were harvested and the total RNA was extracted using the RNAmini Kit (Qiagen, Hilden, Germany). cDNA was generated with cDNA Synthesis Supermix Kit (TransGen Biotech). Quantitative RT-PCR (qPCR) was performed using Hieff® qPCR SYBR Green Master Mix (Yeasen, Shanghai, China) by LightCycler480 (Roche, Basel, Switzerland). GAPDH was used as an internal control. The primers used in qPCR were listed in Table S2.
Cytokine enzyme-linked immunosorbent assay (ELISA)
Effector cells (AKO and P4 CAR-T cells) were cocultured with CRL5826 cells at a 1:1 ratio (2 × 104 cells each). After 3 d, supernatants were harvested and cytokines (IFN-γ and IL-2) produced by effector cells were measured by ELISA Kits (Biolegend, San Diego, California, USA).
Flow cytometry
Harvested cells were stained for 20 min in the dark at room temperature, washed twice with PBS, and analyzed using CytoFLEX (Beckman Coulter Inc., Breia, California, USA). The antibodies were used for flow cytometric analysis where indicated: LAG3-PE (BD Bioscience, 565616, Franklin lakes, New Jersey, USA); CD3-Pacific blue (300329), CD4-APC (317416), CD8-APC (301014), CD8-Brilliant Violet (301035), PD1-PE (329906), TIM3-APC (345012) were all from Biolegend.
Repetitive tumor challenge assay
CAR-T cells were cocultured with CRL5826 cells at a 2:1 ratio, in the presence or absence of CADO or CGS. Every 2 d, CAR-T cells were counted and new tumor cells were added at a constant effector to target ratio 2:1, until CAR-T cells could not lyse tumor cells. The CAR-T cells were collected in the last round and analyzed by FACS to detect the expression of checkpoint genes.
Animal model and in vivo CAR-T cell function detection
Six-week-old female NOD-Prkdcscid Il2rgnull (NPG) mice engrafted with CRL5826 cells or with pancreatic carcinoma PDX (Vitalstar, Beijing, China) were randomly divided into three groups: PBS, AKO, and P4 (n = 5 mice per group), when the tumors were between 300 and 400 mm3 in volume. CAR-T cells were administrated intratumorally twice with a 1-week interval at 5 × 106 cells/mouse (CAR+ was 50%). Tumor sizes were monitored weekly. Mice peripheral blood was collected, and the proportion of CD4 positive and CD8 positive cells was analyzed.
Study approval
All experiments involving animals were approved by Animal Ethic Committee of Institute of Zoology, Chinese Academy of Sciences.
Statistics
Statistical analyses were performed and graphed with Graph-Pad Prism 7 (GraphPad Software Inc., La Jolla, California, USA). Unpaired Student’s t-test and ANOVA with Tukey’s multiple-comparisons test were applied. Data were represented as mean ± s.d.
Author contributions
NL and HYW designed the study, planned the experiments, and wrote the manuscript. NL and TH performed the experiments. NT and CC did the mouse experiment. XFW and WDH edited the manuscript.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download ()Acknowledgments
We are grateful to Wen Sun for editing the manuscript, Yi Yang (Beijing Cord Blood Bank) for her help in preparing the cord blood samples.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Kochenderfer JN, Rosenberg SA. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol. 2013;10(5):267–8. doi:10.1038/nrclinonc.2013.46.
- Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell. 2017;168(4):724–740. doi:10.1016/j.cell.2017.01.016.
- Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. doi:10.1126/scitranslmed.3008226.
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi:10.1056/NEJMoa1407222.
- Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi:10.1056/NEJMoa1707447.
- Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, Brogdon JL, Pruteanu-Malinici I, Bhoj V, Landsburg D, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. 2017;377(26):2545–2554. doi:10.1056/NEJMoa1708566.
- Kosti P, Maher J, Arnold JN. Perspectives on chimeric antigen receptor T-cell immunotherapy for solid tumors. Front Immunol. 2018;9:1104. doi:10.3389/fimmu.2018.01104.
- O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399):eaaa0984. doi:10.1126/scitranslmed.aaa0984.
- Beatty GL, O’Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, Kulikovskaya IM, Soulen MC, McGarvey M, Nelson AM, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology. 2018;155(1):29–32. doi:10.1053/j.gastro.2018.03.029
- Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348(6230):74–80. doi:10.1126/science.aaa6204.
- Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol. 2015;15(11):669–682. doi:10.1038/nri3902.
- Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. Immunology. 2010;129:474–481. doi:10.1111/j.1365-2567.2010.03255.x.
- Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. 2017;276(1):121–144. doi:10.1111/imr.12528.
- Robeva AS, Woodard RL, Jin X, Gao Z, Bhattacharya S, Taylor HE, Rosin DL, Linden J. Molecular characterization of recombinant human adenosine receptors. Drug Dev Res. 1996;39(3‐4:243–252. doi:10.1002/(SICI)1098-2299(199611/12)39:3/4<243::AID-DDR3>3.0.CO;2-R.
- Ohta A, Ohta AM. A2A adenosine receptor may allow expansion of T cells lacking effector functions in extracellular adenosine-rich microenvironments. J Immunol. 2009;183(9):5487–5493. doi:10.4049/jimmunol.0901247.
- Zarek PE, Huang C-T, Lutz ER, Kowalski J, Horton MR, Linden J, Drake CG, Powell JD. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood. 2008;111(1):251–259. doi:10.1182/blood-2007-03-081646.
- Emens L, Powderly J, Fong L, Brody J, Forde P, Hellmann M, Hughes B, Kummar S, Loi S, Luke J. Abstract CT119: CPI-444, an oral adenosine A2a receptor (A2aR) antagonist, demonstrates clinical activity in patients with advanced solid tumors. Cancer Res. 2017;77(13 Supplement):CT119–CT119
- Waickman AT, Alme A, Senaldi L, Zarek PE, Horton M, Powell JD. Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol Immunother. 2012;61(6):917–26. doi:10.1007/s00262-011-1155-7.
- Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MKK, Huang X, Caldwell S, Liu K, Smith P, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci. 2006;103(35):13132–13137. doi:10.1073/pnas.0605251103.
- Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, Davenport AJ, John LB, Mardiana S, Slaney CY, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. 2017;127(3):929–941. doi:10.1172/JCI89455.
- Bergan L, Gross JA, Nevin B, Urban N, Scholler N. Development and in vitro validation of anti-mesothelin biobodies that prevent CA125/Mesothelin-dependent cell attachment. Cancer Lett. 2007;255(2):263–274. doi:10.1016/j.canlet.2007.04.012.
- Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, Wei X-F, Han W, Wang H. TGFβ inhibition via CRISPR promotes the long-term efficacy of CAR-T cells against solid tumors. JCI Insight. 2020;5(4):e133977. doi:10.1172/jci.insight.133977.
- Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17(1):148. doi:10.1186/s13059-016-1012-2.
- Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42(22). e168-e168. doi:10.1093/nar/gku936.
- Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–499. doi:10.1038/ni.2035.
- Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–499. doi:10.1038/nri3862.
- Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110(7):993–1002. doi:10.1172/JCI0215337.
- Vijayan D, Young A, Teng Michele W.L., Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. 2017;17(12):709–724. doi:10.1038/nrc.2017.86.
- Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, Ittmann MM, Marchetti D, Dotti G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21(5):524. doi:10.1038/nm.3833.
- Kloss CC, Lee J, Zhang A, Chen F., Melenhorst JJ, Lacey SF, Maus MV, Fraietta JA, Zhao Y, June CH. Dominant negative TGFβ receptor enhances PSMA targeted human CAR T cell proliferation and augments tumor eradication in prostate cancer. Mol Ther. 2018;26(7):1855–1866. doi:10.1016/j.ymthe.2018.05.003.
- Liu H, Xu Y, Xiang J, Long L, Green S, Yang Z, Zimdahl B, Lu J, Cheng N, Horan LH, et al. Targeting alpha-fetoprotein (AFP)–MHC complex with CAR T-cell therapy for liver cancer. Clin Cancer Res. 2017;23(2):478–488. doi:10.1158/1078-0432.CCR-16-1203.
- Liu X, Zhang Y, Cheng C, Cheng AW, Zhang X, Li N, Xia C, Wei X, Liu X, Wang H, et al. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017;27(1):154–157. doi:10.1038/cr.2016.142.
- Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23(9):2255–2266. doi:10.1158/1078-0432.CCR-16-1300.
- Zhang X, Cheng C, Sun W, Wang H. Engineering T cells using CRISPR/Cas9 for cancer therapy. Methods Mol Biol. 2020;2115:419–433. doi:10.1007/978-1-0716-0290-4_23.
- Zhang Y, Mu W, Wang H. Gene editing in T cell therapy. J Genet Genomics. 2017;44(9):415–422. doi:10.1016/j.jgg.2017.09.002.
- Morote-Garcia JC, Rosenberger P, Kuhlicke J, Eltzschig HK. HIF-1–dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood J Am Soc Hematol. 2008;111(12):5571–5580. doi:10.1182/blood-2007-11-126763.
- Sitkovsky MV, Lukashev D, Apasov S, Kojima H, Koshiba M, Caldwell C, Ohta A, Thiel M. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol. 2004;22:657–682. doi:10.1146/annurev.immunol.22.012703.104731.
- Wilson JM, Ross WG, Agbai ON, Frazier R, Figler RA, Rieger J, Linden J, Ernst PB. The A2B adenosine receptor impairs the maturation and immunogenicity of dendritic cells. J Immunol. 2009;182(8):4616–4623. doi:10.4049/jimmunol.0801279.