
ABSTRACT
Therapeutic cancer vaccines aim to induce an effective immune response against cancer, and the effectiveness of these vaccines is influenced by the choice of immunogen, vaccine type, and immunization strategy. Although treatment with cancer vaccines can improve tumor burden and survival, in most animal studies, it is challenging to achieve a complete response against tumor growth and recurrence, without the use of other therapies in combination. Here, we present a novel approach where dual antigens (survivin and MUC1) are co-targeted using three DNA vaccines, followed by a single booster of a recombinant modified vaccinia Ankara (MVA) vaccine. This heterologous vaccination strategy induced higher levels of interferon (IFN)-γ-secretion and stronger antigen-specific T-cell responses than those induced individually by the DNA vaccines and the MVA vaccine in mice. This strategy also increased the number of active tumor-infiltrating T cells that efficiently inhibit tumor growth in tumor-bearing mice. Heterologous DNA prime-MVA boost immunization was capable of inducing a robust antigen-specific immune-memory, as seen from the resistance to subsequent survivin- and MUC1-expressing tumors. Moreover, the therapeutic effects of DNA prime-MVA boost and DNA prime-adenovirus boost strategies were compared. DNA prime-MVA boost immunization performed better, as indicated by the T effector ratio and the induction of Th1 immunity. This study provides the basis for the use of heterologous DNA prime-MVA boost vaccination regime targeting two antigens simultaneously as a promising immunotherapeutic strategy against cancer.
Introduction
Immunotherapies, including immune checkpoint blockade, oncolytic viruses, and cancer vaccines, have emerged as promising therapeutic approaches for cancer.Citation1,Citation2 Immune checkpoint inhibitors,Citation3,Citation4 represented by inhibitors against cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD1), have been effective in improving the activity of intra-tumoral CD8 T cells for better outcomes in some cancer types. Oncolytic virus immunotherapy, which employs native or engineered viruses that selectively replicate in and kill cancer cells, can promote anti-tumor immunity through the release of antigens from the lysed cancer cells.Citation5,Citation6 The most well-known oncolytic virus, Talimogene laherparepvec (T-VEC),Citation7 is a modified oncolytic herpes simplex virus type 1 armed with cytokine GM-CSF. T-VEC has been approved by the U.S. Food and Drug Administration (FDA) for the treatment of advanced melanoma. Cancer vaccines are designed to prompt the immune system against cancer, and they can be divided into prophylactic and therapeutic vaccines. Prophylactic vaccines usually prevent the infection by oncogenic viruses (hepatitis B, human papillomavirus, etc.),Citation8 whereas, therapeutic vaccines harness the immune system to eliminate existing tumors.Citation8 Cancer vaccines take preponderance because they induce specific anti-tumor immunity for evasive tumors that generally escape immunosurveillance.Citation9,Citation10 Numerous types of cancer vaccines, such as PROVENGE,Citation11 an FDA-approved dendritic cell vaccine for prostate cancer, have been developed. However, these vaccines show varying success in preclinical and clinical studies.Citation10
The choice of antigen targets is a key consideration in designing effective cancer vaccines. MUC1,Citation12–14 a transmembrane glycoprotein, and survivin, an important member of the family of inhibitors of apoptosis proteins,Citation15 are deemed as ideal tumor antigens.Citation16–18 Various vaccines targeting survivin or MUC1, which exhibit immunogenicity and tumor resistance in animals, have been developed.Citation18–21 Considering the heterogeneity of tumor cells, vaccines using a combination of multiple tumor antigens broadens the target spectrum for activated cytotoxic T lymphocytes in target cells.Citation22–24 In our previous studies, DNA vaccines co-targeting survivin and MUC1, administered using an adenovirus vector, exhibited improved therapeutic efficacy, compared with vaccines containing a single antigen.Citation25,Citation26
Amongst various approaches toward the development of improved cancer vaccines, gene-based vaccines, such as DNA, mRNA, and virus vector-based vaccines that are able to induce antigen-specific cellular immunity in vivo, are being explored. DNA vaccines offer numerous advantages over the others as they can readily incorporate multiple genes, are stable, and are easy to engineer.Citation27 However, without the aid of electroporation or vector-mediated delivery, DNA or mRNA vaccines have limited cell transfection efficiency in their naked forms.Citation28–32 In contrast, virus vector-based vaccines intrinsically mediate highly efficient transduction of the antigen gene. This contributes to the endogenous expression of antigens for induction of adaptive cellular immunity.Citation33 In many studies, adenovirus and modified vaccinia Ankara (MVA)-based cancer vaccines have been evaluated and proven to be immunogenic and safe.Citation34 We have developed an adenovirus-based cancer vaccine targeting MUC1 and surviving, and demonstrated its immunogenicity and therapeutic efficacy in mouse models.Citation25,Citation26 MVA, a highly attenuated vaccine vector with the authentic confirmation of virus-like particles (VLPs), is popular for immunogenic purposes due to its ability to accommodate large antigen sequences in the genome, high safety, and natural adjuvant effect in activating innate immunity to promote antigen presentation.Citation34 This study aimed to develop an MVA vaccine targeting dual antigens and evaluated its immunogenicity and therapeutic efficacy.
In addition to the target immunogen and the type of vaccine, the vaccination regime also affects the outcome of immunization. Multiple immunizations in the form of prime-boost is usually required for effective immunoreactions.Citation35 In many cases, a heterologous prime-boost administered with different types of vaccines containing the same antigens was more immunogenic than a homologous prime-boost that was repeatedly administered with the same type of vaccine. In combinations of heterologous prime-boost strategies, a DNA prime followed by boosting with viral vectors have enhanced immune responses to malaria,Citation36 viruses,Citation37,Citation38 and cancersCitation25,Citation26,Citation39,Citation40.
Here, we aimed to develop a vaccine-based therapeutic strategy against cancer, the design of which involved the selection of multi-antigens, delivery using a viral vector, and exploring heterologous prime-boost vaccinations. A vaccine targeting both survivin and MUC1 was constructed using MVA. The homologous immunization regimes were evaluated to understand the efficacy of the individual MVA vaccines to induce a robust anti-tumor immunity in mice. To further enhance the tumor inhibition efficacy of dual antigens, a vaccination regimen of three DNA vaccine administrations, followed by single recombinant MVA boost was employed. The immunogenicity and therapeutic efficacy of DNA prime-MVA boost strategy was explored and compared with the homologous immunization strategies and a DNA prime-adenovirus boost strategy. Our findings established the advantage of DNA prime-MVA boost strategy over the others to induce antigen-specific immune memory to resist cancer recurrence.
Materials and methods
Mice and cell lines
Female C57/BL/6 mice, 6–8 weeks old (Beijing Huafukang Biology Technology Co., Ltd., China), were used in this study. All animal procedures were conducted in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University Committee on the Use and Care of Animals of Jilin University of China.
B16 and BHK-21 tk-ts13 cell lines were purchased from ATCC Cell Bank, and Lewis (GF123) cell line was purchased from Shanghai Gefan Biotechnology Co., Ltd., China.
MSf+ Lewis and MSf+ B16 cell lines stably expressing MUC1 and survivin were generated in our lab.Citation25,Citation26 Briefly, B16 cells or Lewis cells were transfected with a GFP-MS plasmid (containing a fusion of G418 resistance, MS, and GFP genes). G418 (final concentration: 0.8–1 mg/mL) was used to select the transfected cells 24 h post-transfection until the majority of cells expressed GFP. The cells were cultured in 96-well plates at single cell per well. Monoclonal cell lines stably expressing MS and GFP were obtained.
Preparation of MVA vaccine armed with dual antigens
An sPD1-MUC1-survivin fusion gene, used in our previous study,Citation20 was inserted into the shuttle vector pSC11M1, and termed as pSC11M1-MS. The sPD1 protein was utilized to improve the cross-presentation of antigens. Homologous recombination was performed in BHK-21tk-ts13 cells for the production of recombinant MVA, according to the standard procedures. In brief, the BHK-21 tk-ts13 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS; Gibco, Thermofisher), infected with WT-MVA at an MOI of 0.05, and transfected with pSC11M1-MS 2 h post-infection. The rMVA was selected by blue-white selection; 5-bromodeoxyuridine was used for thymidine kinase (TK) selection and recombinant MVA amplification. Zonal sucrose gradient centrifugation was used for purification of rMVA. The rMVA titer was determined via immunostaining with an anti-vaccinia antibody (Fitzgerald) and horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin G (IgG) (Jackson ImmunoResearch).
Antigen gene expression of MVA vaccine
BHK21 tk-ts13 cells were cultured in DMEM containing 10% FBS (Gibco) and exposed to rMVA or WT-MVA at an MOI of 10. At 24 h post-infection, cells were harvested and gene expression was detected via western blotting and immunocytochemical staining using primary antibodies of survivin (1:1000, Novus-Biologicals) and MUC1 VNTR (1:1000, BD Pharmingen). For western blotting, HRP-labeled goat anti-rabbit IgG (Jackson ImmunoResearch) and HRP-labeled rabbit anti-mouse IgG (Jackson ImmunoResearch) were used as the secondary antibody. For immunocytochemical staining, CoraLite488-conjugated goat anti-rabbit IgG (Proteintech, 1:500) and Cy5-labeled goat anti-mouse IgG (Bioss) were used.
Mice and immunization for immunogenicity evaluation in C57/BL/6 mice
MVA vaccine was administered at 1-week interval twice or three times, when used alone. DNA vaccine was administered for three times in a week, when used alone. For heterologous prime-boost group, MVA vaccine was administered 1-week post the three DNA vaccinations. All vaccines were administered through intramuscular injection into the tibialis anterior muscles of both hind limbs. DNA vaccine was injected at a concentration of 100 μg per mouse (50 μg in 50 μL PBS for each limb) at each vaccination, and MVA vaccine was injected at 5 × 107 plaque-forming units (PFUs) (2.5 × 107 PFUs in 50 μL PBS for each limb) per mouse per time.
ELISpot and cytotoxicity assays
IFN-γ molecules released by antigen-stimulated splenocytes were detected using an ELISpot kit ((BD Biosciences) as previously described.Citation41
Cytotoxicity assays were performed in accordance with previously published procedures.Citation20 In brief, MSf+ Lewis and Lewis cells as target cells were labeled with different concentrations of CFSE fluorescent dye. A total of 5 × 105 splenocytes isolated from immunized mice were incubated at different effector-to-target (E:T) ratios with the target cells for 6–8 h at 37 C and 5% CO2. The cytotoxicity was analyzed using flow cytometry, and the antigen-specific lethality was calculated according to the formula: specific cytotoxicity = [1 − (MSf+ Lewis cells/unloaded cells from the immunized group)/(Lewis cells/unloaded cells from the naïve group)] × 100%.
Analysis of T cells in splenocytes after stimulations
Splenocytes were either stimulated with survivin or MUC1 protein for 12 h. Following incubation, the cells were stained with the following surface antibodies: APC anti-mouse CD8a (Biolegend), PE-Cy7 anti-mouse CD4 (Biolegend). Cells were then permeabilized and washed with the Cytofix/Cytoperm kit (BD Biosciences) according to manufacturer’s instructions. Intracellular Ki67 were subsequently stained with PE anti-mouse Ki67 (Biolegend). For central memory T cells (TCM) and effector memory T (TEM) cells, in addition to APC anti-mouse CD8a (Biolegend), PE anti-mouse CD44 (Biolegend) and PE-Cy7 anti-mouse CD62L (Biolegend) were used for cell surface staining. After staining, cells were washed, fixed, and analyzed using flow cytometry.
Detection of specific antibodies using ELISA
Serum from mice was diluted with 1× PBS at a ratio of 1 to 50 to detect antibodies against MUC1 and survivin using ELISA performed according to previously described methods.Citation25 MUC1 and survivin proteins purified from recombinant E. coli BL21 were coated at 100 ng per well overnight. The wells were blocked using BSA, and the cells incubated with primary antibody for 2 h followed by incubation with HRP-labeled secondary antibodies (Jackson ImmunoResearch) for 1 h and then TMB was added. Sulfuric acid (2 M) was used to stop the reaction, and the OD450 values were determined.
Detection of binding antibody and neutralizing antibodies against MVA vector
Mice were intramuscularly (i.m.) injected with MVA vaccine thrice at one-week intervals. 5 × 107 PFUs MVA vaccine was injected per mouse per time (2.5 × 107 PFUs in 50 μL PBS for each limb). The day before each vaccination, blood was drawn from the mice, and serum was prepared.
ELISA was used for the detection of binding antibodies. MVA vectors were coated in the plate at 1 × 106 PFUs per well and was incubated at different dilutions of the serum for 2 h followed by HRP-labeled secondary antibodies (Jackson ImmunoResearch) for 1 h. After reaction with TMB and sulfuric acid (2 M) termination, OD450 values were measured.
Cell viability detection was used for screening the neutralizing antibodies against MVA vector in mouse serum. BHK 21 tk-ts13 cells were plated in 96-well plates at 5000 cells per well and cultured for 24 h. The MVA vectors were incubated with varying dilutions of serum, at 37 C for 30 min, and then were added to the wells. The medium was changed to DMEM with 2% FBS at 2 h post-virus infection, and the cells were cultured for another 48 h. Cell viability was detected using MTT assay. The cell viability ratio (%) was calculated according to the following formula: [(absorbance of experimental group with virus infection− background absorbance)/(absorbance of control group without virus infection − background absorbance)] × 100%.
Therapeutic experiments in tumor-bearing mice
MSf+ Lewis lung cancer cells (1 × 105) were injected subcutaneously into the right back flank of the mice at day 0. Mice were vaccinated from day 4. The usage of vaccines and vaccination intervals were set at the same levels as the immunogenicity evaluation experiment. Tumor size was measured every two days, and the tumor volume was calculated as (length × width2)/2. The tumor inhibition rate (%) was calculated as (average tumor volume of PBS group – average tumor volume of experimental group)/average tumor volume of PBS group × 100%.
Analysis of immune cells in tumor
Tumor tissues, excised from euthanized mice, were minced and digested using liberase (Roche) for 2 h. The cells were washed and counted, followed by extracellular staining with antibodies, such as APC anti-mouse Gr-1 (Biolegend), PE anti-mouse CD11b (Biolegend), FITC anti-mouse F4/80 (Biolegend), and APC anti-mouse CD206 (Biolegend), to analyze MDSCs and TAMs in the tumor. For analysis of the tumor-infiltrated proliferative T cells, the cells were washed, surface-stained using PE-Cy7 anti-mouse CD4 (Biolegend), and APC anti-mouse CD8a (Biolegend), fixed, and permeabilized, followed by intracellular staining using PE anti-mouse Ki67 (Biolegend). Cells were washed and analyzed using flow cytometry. Detection of Tregs (regulatory T cells) was performed using Mouse Regulatory T Cell Staining Kit#1 (eBioscience), according to manufacturer’s instructions.
Quantification of intra-tumoral cytokine expression by qRT-PCR
Total RNA extraction from tumors was performed using an RNeasy kit (Qiagen), and the mRNA was reverse transcribed to cDNA using the PrimeScript 1st Strand cDNA Synthesis Kit (Takara Biotechnology Co.) The mRNA levels were quantified using qRT-PCR. The primers used to detect IL-2, IL-4, IL-6, IL-10, IFN-γ, TNF-α, and GzmB mRNA expression and the procedure for qRT-PCR were as described previously.Citation42 GAPDH was used as the internal reference.
Statistical analysis
Data were analyzed using an unpaired t-test or one-way analysis of variance (ANOVA) followed by the least significant difference (LSD) analysis. Analyses were performed using SPSS (IBM SPSS Statistics 20). The results are expressed as the mean ± standard error and are considered significant at P < .05.
Results
Gene expression and immunogenicity of MVA vaccine
The recombinant MVA vector (rMVA) incorporated with the sequences corresponding to survivin and MUC1 was constructed for a dual-gene cancer vaccine (). To verify the expression of these antigens, survivin, and MUC1 proteins were detected in rMVA-infected BHK tk-ts13 cells using immunocytochemical staining and western blotting analysis at 24 h post-infection. rMVA mediated the expression of sPD1-MUC1-survivin, whereas the wild-type MVA without fusion gene insertion did not as observed from the confocal images (, Figure S1) and flow cytometry analysis result (, Figure S2). MUC1 and survivin fusion protein expression were also detected on the western blots (), using specific antibodies. Multiple protein bands were observed on the western blot and the primary band appeared at a higher molecular weight than expected. This could be explained by the glycosylation of the MUC1 protein. The rMVA coded sPD1-MUC1-survivin fusion protein should be in the glycosylated form in the MUC1 region and, therefore, migrate at a higher molecular weight.
Figure 1. Modified vaccinia Ankara (MVA) vaccine-mediated immunogen expressions and the immunogenicity. (a) Schematic of modified vaccinia Ankara (MVA) vaccine used in this study. The MVA vaccine is armed with a fusion gene of sPD1-MUC1-S8. The sPD1 protein is used to enhance the cross-presentation of antigens; MUC1 contained 33 repeats of VNTR; S8 is a deleted form of survivin without the first seven amino acids. (b-c) Confocal images and flow cytometry analysis of MUC1 and survivin expression after intracellular immunostaining and BHK tk-ts13 cells at 24 h post-MVA infection. Scale bar represents 50 µm. (d) Western blotting analysis of MUC1 and survival rates in BHK tk-ts13 cells at 24 h post-MVA infection. (e) Immunization regime illustration; total of four to five mice were in each group. (f, g) Representative images of ELISpot and quantification of ELISpot SFUs in groups. (h) Cytotoxicity evaluation of MVA vaccine-immunized mice (E: T, effector: target cells ratio). N.s., nonspecific protein; M: MUC1 protein; S, survivin protein. Groups drawn in colors: Vehicle (PBS; black), 2MVA-1 (red), 2MVA-2 (brown), 2MVA-3 (pink), 3MVA (blue). One-way ANOVA followed by LSD analysis was performed to analyze the significant differences between groups. *P < .05; **P < .01; ***P < .001
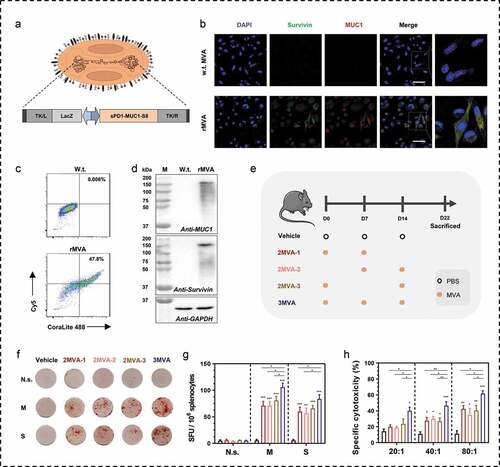
To determine if MVA vaccine is immunogenic in mice, C57/BL mice were vaccinated using two-needle or three-needle strategies (). The 2MVA-1 group was vaccinated at the same starting time as the 3MVA group, whereas the 2MVA-2 group was immunized at the same ending time as the 3MVA group. Both MVA groups were vaccinated at 1-week intervals, whereas the 2MVA-3 group was vaccinated with MVA twice at 2-week intervals. Interferon gamma (IFNγ) ELISpots (–g, Figure S3) was performed using proteins or peptides matched to the antigens. The group that received the three-needle administration of the rMVA indicated more spot-forming units (SFUs) toward dual antigens stimulated by either proteins or peptides compared with those subjected to the two-needle strategies (2MVA-1, 2MVA-2, 2MVA-3). In addition, three-needle administration of rMVA induced the highest specific cytotoxicity toward antigen-expressing cancer cells at all E:T ratios in CTL assay ().
Anti-tumor efficacy of MVA vaccine administered as homologous prime-boost
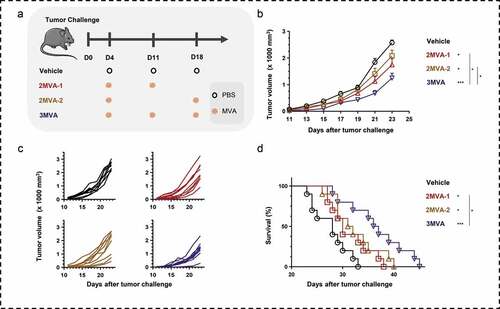
The therapeutic efficacy of MVA vaccine was evaluated in tumor-bearing mice, using two-needle (2MVA-1, 2MVA-2) or three-needle (3MVA) homologous vaccination strategies (). Mice in both groups, vaccinated with either rMVA twice or thrice, had reduced tumor growth (,c), compared to the PBS-treated group. The specific cellular immune response against immunogens was induced by the recombinant MVA and not by MVA vector without MUC1 and survivin (Figure S10C, D). The tumor inhibition rate of 2MVA-1 and 2MVA-2 strategies were 21.72% and 22.78%, respectively, whereas that of the 3MVA strategy was 42.64%. Meanwhile, the life spans of the mice in the 2MVA-1, 2MVA-2, and 3MVA groups were significantly prolonged, especially the 3MVA group ().
Figure 2. Therapeutic efficacy of modified vaccinia Ankara (MVA) vaccine. (a) Schematic of the therapeutic regime. A total of 1 × 105 MSf+ Lewis cells were injected subcutaneously on the right back of C57/BL/6 mice on day 0, and vaccinations began 4 days after tumor inoculation. (b) Tumor growth curve in groups; each group involved nine to ten mice. (c) Tumor growth curve of each mice in groups. (d) Survival curve; each group involved ten mice. Groups drawn in colors: Vehicle (PBS; black), 2MVA-1 (red), 2MVA-2 (brown), 2MVA-3 (pink), 3MVA (blue). One-way ANOVA followed by LSD analysis was performed to analyze the significant differences between groups. *P < .05; **P < .01; ***P < .001
Neutralizing antibody against MVA vector is rapidly generated after MVA vaccination
Neutralizing antibody against MVA vector will restrict the efficiency of boosting.Citation43 To explore whether the neutralizing antibody is generated rapidly, we detected binding antibodies and neutralizing antibodies in serum at four time points: one day before the first MVA immunization (day 1), six or seven days post the immunizations (day 6, day 13, and day 21). The experimental design is shown in .
Figure 3. Binding antibodies and neutralizing antibodies against MVA vectors after immunizations. (a) Schematic of rMVA vaccination regime and time points of blood collection. (b) Serum antibody against MVA vector was detected by ELISA. The serum was diluted from 1:100 to 1:12500. (c) Cell viability of BHK tk-ts13 cells at 48 h post-infection of serum-incubated rMVA. The serum was used at the dilution of 1:40 to 1:2560. (d) Representative morphology images of BHK21 tk-ts13 cells at 48 h post-infection of serum-incubated rMVA. Groups drawn in colors: serum before immunizations (Day −1; black), serum after the first immunization (Day 6; blue), serum after the second immunization (Day 13; red), serum after the third immunization (Day 21; brown). One-way ANOVA followed by LSD analysis was performed to analyze the significant differences between groups. Scale bar represents 50 μm. *P < .05; **P < .01; ***P < .001
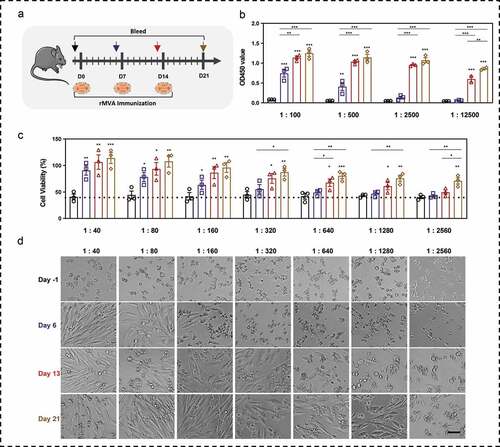
Binding antibody () against MVA vector was generated after the first immunization, which could be detected using ELISA until a serum dilution of 1: 2500. The antibody levels increased with the number and time after MVA injections. There was a significant increase in OD450 values in the serum at 1:12500 dilution, after the second and third immunizations.. Since MVA is capable of replicating in BHK21 tk-ts13 cells and inducing cell death, we analyzed the effect of serum neutralizing antibodies through measuring cell viability after infection with serum-incubated MVA. Based on the cell viability () and cellular morphology () assays, we observed that the cell viability could be significantly improved by serum collected after the first immunization up to a dilution of 1:160, indicating that neutralizing antibodies were generated in serum with even a one-time MVA vaccination. The serum collected after the second immunization significantly improved cell viability by neutralizing MVA up to a dilution of 1: 1280, whereas the serum collected after the third immunization significantly rescued the cytotoxicity of MVA infection up to a dilution of 1:2560.
DNA prime and MVA boost enhances immunity toward antigens
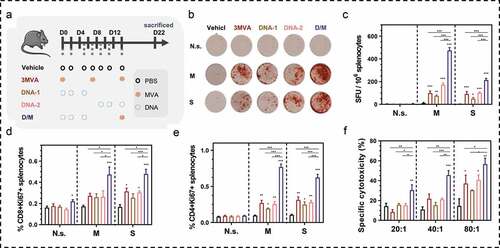
An approach to avoid the influence of neutralizing antibodies against viral vectors is to use a heterologous prime-boost strategy, instead of the homologous prime-boost. Heterologous prime-boost has shown to be more immunogenic than homologous prime-boost.Citation35 Here, we designed a prime-boost strategy with a DNA vaccine and the MVA vaccine containing the same antigens and evaluated the immunogenicity of the heterologous prime-boost strategy in C57/BL/6 mice (). Wild-type MVA vector and DNA vector without the antigens, MUC1, and survivin did not induce antigen-specific cellular immune response (Figure S10). PBS was set as a control to minimize variation between the groups in this study. The ELISpot assay (,c, S4) was performed to evaluate the IFN-γ secretion from splenocytes after antigen-stimulation, as an indication of cellular immune response in mice. With MUC1, the D/M strategy induced an approximately 7.2 times increase in SFUs compared to MVA vaccination strategy (homologous vaccinations using rMVA alone), and approximately 4.1 to 9.6 times increase in SFUs compared to DNA vaccination strategy (homologous vaccinations using DNA alone). With survivin, the D/M strategy induced approximately 2.3 times increase in SFUs compared to MVA vaccination strategy, and approximately 2.0 to 4.2 times increase in SFUs compared to DNA vaccination strategy. Moreover, the proliferative CD8+ T (CD8+Ki67+) and proliferative CD4+ T (CD4+Ki67+) cells significantly increased in the splenocytes of immunized mice following survivin or MUC1 stimulation, with the largest fold-increase in the D/M group, whereas no changes were induced in response to the vehicle-treated mice (,e). CTL assay was also performed to evaluate the specific cytotoxicity toward MUC1 and survivin expressing cells (). Although significant specific cytotoxicity was observed at all E:T ratios in the D/M group, it was observed only at the 80:1 of E:T ratio in the other homologous groups. Therefore, D/M strategy induced the most efficient specific cytotoxicity. These results show the effective induction efficiency of cellular immune DNA prime-MVA boost regime.
Figure 4. Immunogenicity evaluation of DNA prime-modified vaccinia Ankara (MVA) boost strategy. (a) Schematic of the vaccination regime; total four to five mice in each group. (b, c) Representative images of ELISpot and quantification of ELISpot SFUs in groups. (d, e) Percentage of proliferative CD4+ T (CD4+Ki67+) or proliferative CD8+ T (CD8+Ki67+) in splenocytes after stimulation with survivin or MUC1 protein. (f) Cytotoxicity evaluation of MVA vaccine-immunized mice (E: T, effector: target cells ratio). N.s., nonspecific protein; M: MUC1 protein; S, survivin protein. Groups drawn in colors: Vehicle (PBS; black), 3MVA (red), DNA-1 (brown), DNA-2 (pink), D/M (blue). One-way ANOVA followed by LSD analysis was performed to analyze the significant differences between groups. *P < .05; **P < .01; ***P < .001
Apart from cellular immune response, antibodies against survivin and MUC1 were analyzed using ELISA. The heterologous vaccinations induced stronger humoral immune response in mice, compared to the homologous strategies, as indicated by the amounts of total antibodies, for both survivin and MUC1 (Figure S5).
The heterologous DNA prime-MVA boost strategy significantly improves therapeutic effects in tumor-bearing mouse model
The therapeutic effect of the DNA prime-MVA boost heterologous strategy was further evaluated in lung tumor-bearing mice (). Tumor growth was inhibited in mice vaccinated with the homologous vaccination strategies. However, the D/M heterologous vaccination strategy reduced the tumor growth most efficiently (,c). In addition to tumor aversion, the survival of tumor-bearing mice was prolonged, especially in the D/M group (). Notably, three mice out of 10 mice in D/M group survived with no tumor at the end of the experiments.
Figure 5. Comparison of therapeutic efficacy among the different heterologous prime-boost strategies. (a) Schematic of the therapeutic regime. 1 × 105 MSf+ Lewis cells were injected subcutaneously on the right back of C57 bl/6 mice on day 0, and vaccinations began 4 days after tumor inoculation. (b) Tumor growth curve in groups; each group included ten mice. (c) Tumor growth curve of each mice in groups. (d) Survival curve; each group involved nine mice. (e–g) Percentage of intra-tumoral proliferative CD4+ T (CD4+Ki67+), proliferative CD8+ T (CD8+Ki67+) and Treg (CD4+CD25+Foxp3+) were analyzed using flow cytometry. (h) The ratio of CD8+Ki67+ T cells to Tregs. (i) Analysis of intra-tumoral MDSC. (j, k) The percentage of MDSC (CD1b+Gr-1+), TAM-M1 (F4/80+CD206−), and TAM-M2 (F4/80+CD206+) was analyzed using flow cytometry. (l, m). Relative mRNA expression of Gzmb and Th1/Th2 cytokines was analyzed using qRT-PCR. Groups drawn in colors: Vehicle (PBS; black), 3MVA (red), DNA-1 (brown), DNA-2 (pink), D/M (blue). One-way ANOVA followed by LSD analysis was performed to analyze the significant differences between groups. *P < .05; **P < .01; ***P < .001
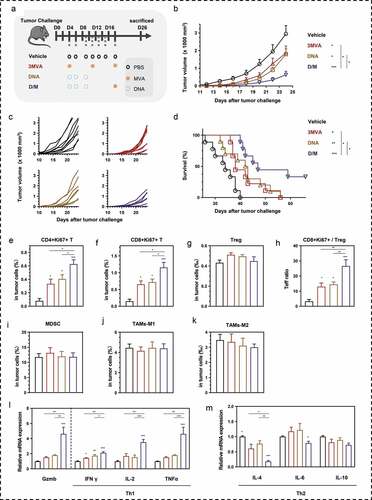
To verify the effects of DNA prime-MVA boost strategy in stimulating cellular response against tumor, we analyzed the tumor-infiltrated immune cells. The number of active proliferative T cell infiltration improved in all vaccinated groups, as indicated by the increase of Ki67+ percentages in CD4+ and CD8+ T cells (,f). Furthermore, vaccine administration did not alter the percentage of Treg in all tumor cells (). Compared with other groups, the T effector (Teff) ratio of active proliferative CD8+ T cells to Tregs in the tumor was the highest in the D/M group (). The number of MDSCs and TAMs in tumors was unaffected in all vaccination strategies (-k).
The increased intra-tumoral mRNA expression of GzmB and Th1 cytokines (IFN-γ, IL2, and TNF-α) and reduced expression of Th2 cytokines (IL-4, IL-6) reflected the ability of DNA prime-MVA boost in mediating a shift from Th2 to Th1 immunity locally in the tumor (,m).
MVA vector-based vaccines have been demonstrated safe in preclinical and clinical studies. Body weight was recorded and hematoxylin-eosin (HE) staining of mouse tissues was performed to analyze the safety of vaccines (Figure S6). There was no change in the body weight. No obvious differences were found between MVA-administrated groups (3MVA, D/M) and other groups (vehicle, DNA) in histopathology.
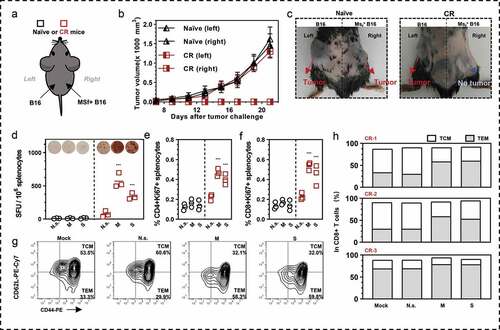
Immunological memory in complete response (CR) mice
Three CR mice from the D/M group were re-challenged with MSf+ Lewis cells at day 90 post-primary tumor induction. For a continuous duration of 25 days, there was no tumorigenesis, indicating that anti-tumor immunological memory have been developed in these mice. To clarify if the immunological memory specific to survivin and MUC1 antigens was induced by our vaccines, we continued to challenge mice with B16 and MSf+ B16 cells at both flanks of the mice (). The third tumor challenge was performed on day 115 post-primary tumor transplant. Three naïve mice were used as control. In naïve mice, both injection of B16 at the left side and MSf+ B16 at the right side caused tumors; however, in the CR mice, only the B16 challenge on the left caused a tumor, with no observable tumor on the right (,c). The CR mice were euthanized and splenocytes were collected to analyze the systemic anti-tumor immunological memory. Nearly 600 SFUs/106 splenocytes of CR mice were stimulated by MUC1, and approximately 340 SFUs were stimulated by survivin (). In addition, the percentage of proliferating T cells (CD4+Ki67+ T or CD8+Ki67+ T) significantly increased following survivin or MUC1 stimulation (,f). We observed abundant antigens specific SFUs following stimulation with survivin and MUC1 peptides (Figure S7). Moreover, the central memory T cells (TCM; CD44+CD62Lhigh) converted to effector memory T cells (TEM; CD44+CD62Llow) following stimulation by antigens (,h).
Figure 6. Antigens specific immune memory analysis in (CR) mice. (a) Three CR mice from D/M immunized group were inoculated with B16 cells (3 × 105 cells) on the left back and MSf+ B16 cells (3 × 105 cells) on the right back, after they survived from secondary MSf+ Lewis tumor attack. (b) Growth curve of tumors on both sides of naïve mice or CR mice. (c) The representative images of tumor formation in CR mice or naïve mice at day 25 post tumor inoculations on both sides. (d) Representative images of ELISpot and quantification of ELISpot SFUs in groups. (e, f) Percentage of proliferative CD4+ T (CD4+Ki67+) or proliferative CD8+ T (CD8+Ki67+) in splenocytes after stimulation with survivin or MUC1 protein. Groups drawn in colors in (a-f): Naïve mice (black), CR mice (red). (g, h) representative results and percentage of TCM and TEM in each CR mice analyzed by flow cytometry; Mock, no stimulation; N.s., nonspecific protein stimulation; M, MUC1 protein stimulation; S, survivin protein stimulation. One-way ANOVA followed by LSD analysis was performed to analyze the significant differences between groups. *P < .05; **P < .01; ***P < .001
Comparison of the therapeutic effects of MVA vaccine and adenovirus-vaccine boosts
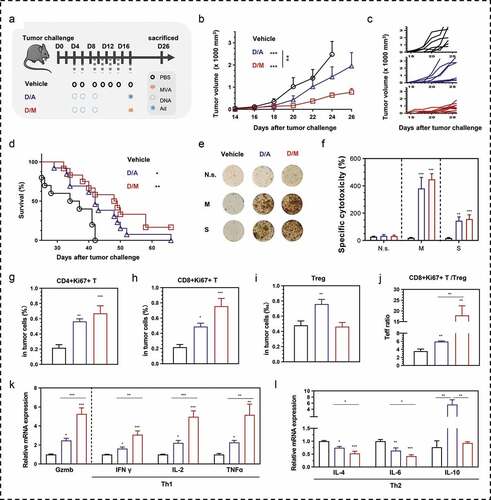
To determine the more effective DNA prime-virus boost heterologous immunization strategy in inducing anti-tumor immunity, we compared between D/M and DNA prime-adenovirus boost (D/A) in mice bearing MSf+ Lewis tumor (). D/M strategy inhibited tumor growth more efficiently than D/A vaccinations (,c). Mice in D/M group exhibited longer median survival (48.5 days) with two CR mice surviving with no tumors, compared to the median survival of D/A (42 days) and vehicle (35.5 days) groups (). To understand observations, an ELISpot assay was used to analyze the antigen-specific cellular immune response in splenocytes. There were no significant differences in SFUs stimulated by MUC1 and survivin protein (,f) or peptides (Figure S8) between the two heterologous groups. Intra-tumoral proliferative CD4+ and CD8+ T cells were more abundant in the tumors of the D/M group than in those of the D/A group (,h). The number of Tregs increased in the tumor of the D/A group compared with that in the vehicle and D/M groups (), and this increase might be the cause of the limited proliferative tumor-infiltrated T cells and the consequent low Teff ratio in the tumors (). The mRNA expression levels in the tumors were analyzed using qRT-PCR (,l). Gzmb and Th1 cytokines exhibited higher expression than Th2 cytokines, including IL-4 and IL-6, in the D/M group compared with that in the D/A group. Compared to the vehicle and D/M groups, the D/A group showed higher expression of IL-10, a cytokine that suppresses anti-tumor cellular immunity. The higher expression of Th1 cytokines and the markedly lower expression of Th2 cytokines indicated a stronger Th1 immunity and weaker Th2 immunity in the tumors of the D/M group, compared to the D/A group.
Figure 7. Comparison of therapeutic efficacy between DNA- modified vaccinia Ankara (D/M) and DNA-adenovirus (D/A) prime-boost strategies. (a) Schematic of the therapeutic regime. 1 × 105 MSf+ Lewis cells were injected subcutaneously on the right back of C57 bl/6 mice on day 0, and vaccinations began 4 days after tumor inoculation. (b) Tumor growth curve in groups; each group involved ten mice. (c) Tumor growth curve of each mice in groups. (d) Survival curve; each group included nine mice. (e, f) Representative images of ELISpot and quantification of ELISpot SFUs in groups. (g–i) Percentage of intra-tumoral proliferative CD4+ T (CD4+Ki67+), proliferative CD8+ T (CD8+Ki67+) and Treg (CD4+CD25+Foxp3+) were analyzed using flow cytometry. (j) The ratio of CD8+Ki67+ T cells to Tregs. (k, l) Relative mRNA expression of Gzmb and Th1/Th2 cytokines were analyzed using qRT-PCR. N.s., nonspecific protein stimulation; M, MUC1 protein stimulation; S, survivin protein stimulation. Groups drawn in colors: Vehicle (PBS; black), D/A (blue), D/M (red). One-way ANOVA followed by LSD analysis was performed to analyze the significant differences between groups. *P < .05; **P < .01; ***P < .001
Discussion
The combined use of multi-antigens in a vaccine may broaden the spectrum of target cells.Citation22–24 Our previous study demonstrated that incorporating survivin and MUC1 VNTRs into a DNA vaccine had a more efficient therapeutic effect than did a vaccine containing a single immunogen.Citation25,Citation26 However, the use of DNA as a vector has its limitations, including low-immunogenicity, which can be improved by delivering antigens using viral vectors.Citation33 Recombinant poxviruses are popular vectors for immunogenic purposes, and the safety of MVA has also been tested in clinical trials.Citation34 Inspired by these results, we constructed a cancer gene vaccine, which is based on an MVA vector that is armed with survivin and MUC1 as immunogens. When only one dose of the MVA vaccine was used, no therapeutic effects were observed in an established mouse model (Figure S9). However, when a homologous prime-boost with a total of two or three vaccinations was administered, the immunogenicity and therapeutic effects were enhanced, especially with three vaccinations (, ).
MVA has high immunogenicity, easily inducing the generation of antibodies against the viral vector.Citation43 In this study, antibodies including neutralizing antibodies against MVA vector were rapidly generated in mice, after only one-time MVA vaccine injection (i.m.) (). In contrast to oncolytic VCAC that can replicate in cancer cells, MVA vaccines cannot replicate in injected human or mouse tissues (muscle or subcutaneous tissues). The efficiency of antigen expression relies on the infectious efficacy of the non-replicating MVA vaccine. Thus, antibodies against viral vector might influence, but not severely alter the therapeutic effects of intra-tumoral replicating oncolytic VCAC.Citation44 However, neutralizing antibodies will possibly attenuate the infectivity of non-replicating MVA vaccines and further reduce antigen expression, when MVA vaccines are repeatedly injected intramuscularly or subcutaneously. In addition, vaccination with MVA vaccine easily induces anti-vector immunity compared to oncolytic VCAC, as VCAC is mainly located in immunosuppressive tumor microenvironment.
We expected MVA vaccine to induce stronger cellular immune response against MUC1 and survivin than DNA vaccine. However, three administrations indicated little differences in inducing immune response (3MVA group vs. DNA-1 or DNA-2 group, –f) and anti-tumor efficacy (3MVA group vs. DNA group, ), when compared to DNA administrations. When MVA vaccines are administered multiple times, the neutralization antibodies may attenuate the effects of subsequent administrations following the first vaccination. In this study, DNA vaccine involved CpG, IL-2 and sPD1Citation25,Citation26 to improve the immunogenicity and was administered via electroporationCitation28. These may explain why the triple use of MVA vaccine showed similar anti-tumor response with DNA vaccine in vivo.
To avoid the influence of neutralization toward homologous MVA prime-boost and further improve the immunogenicity toward dual antigens, a heterologous prime-boost regime can be considered as an ideal approach. DNA prime-virus boost has been widely accepted as an effective immunization strategy. A rapid immunization of DNA (thrice) was performed in a week, followed by a single MVA boost (). Compared to the homologous prime-boost strategies, the combined use of DNA vaccine and MVA vaccine significantly improved the cellular immune response toward dual antigens in mice (–f), as indicated by the results of ELISpot, cytotoxicity assays, and T cell proliferation assays, following antigen stimulation. Moreover, as observed from the levels of antibodies, the D/M strategy enhanced the humoral immune response (Figure S4). As expected, this strategy efficiently inhibited tumor growth and prolonged the survival of mice (–d) through increasing the intra-tumoral proliferative CD4+ T and CD8+ T cells for a higher Teff (–h).
It is expected that immune memory, against tumor recurrence, will be generated in response to immunotherapies, but the results usually disappoint researchers. In our experimental sets, however, three mice (,d) and two mice (,d) survived with CR, following the administration of the D/M strategy vaccination to target survivin and MUC1 immunogens. When MSf+ Lewis cells were injected subcutaneously for a second time, no tumor formations were observed. Thus, we concluded that these CR mice had obtained immune memory. To verify that the immune memory was antigen (survivin and MUC1) specific, the three CR mice in a prior experiment were challenged with B16 tumor cells on the left back and MSf+ B16 cells on the right back (). Antigen-overexpressed tumor was eradicated, whereas B16 could survive in CR mice (, S6). In addition, B16 tumors grew more slowly in CR mice than in naïve mice, possibly due to the specific memory of human survivin and MUC1, inducing a response to the mouse antigens that are naturally expressed in B16 cells. The number of proliferative CD4+ T and CD8+ T cells in splenocytes significantly increased (–f), causing TCM to transform into TEM (–h) following survivin or MUC1 protein stimulation. To observe the anti-tumor effects, D/A, a commonly employed heterologous prime-boost strategy, was compared with D/M strategies (). DNA vaccine was administered three times in a week, and MVA- or adenovirus-vaccine armed the same antigens were used as the booster dose. D/M performed better in tumor inhibition than in D/A (–d). Nearly no differences in SFU formation were observed between D/M and D/A groups, based on the ELISpot results (–f), reflecting the lack of peripheral (in spleen) cellular immune response. In addition, in the tumors, we observed lower expression of certain anti-tumor cellular immunity suppressors (Tregs and IL-10 expression) and a higher Teff ratio in the D/M group, compared to the D/A group.
This study mainly presented a novel three-needle DNA vaccine strategy that is boosted with a single-needle MVA vaccine for the efficient targeting of dual tumor antigens of survivin and MUC1 to inhibit tumor growth and recurrence. The D/M strategy was shown to be superior to homologous prime-boost strategies and the D/A strategy, thereby, providing a reference for future studies on cancer vaccines. For example, neoantigens targeted and personalized cancer vaccineCitation45 may be better at inducing anti-tumor immunity, if it is designed and used based on the D/M strategy. On the basis of the robust immunoreactivity toward dual antigens induced by D/M immunization strategy, this vaccine approach could be potentially combined with other therapies in the future, such as immune checkpoint inhibitors,Citation46,Citation47 chemotherapy,Citation48,Citation49 or radiotherapy,Citation50,Citation51 that could further improve the intratumoral infiltration of T cells and enhance the activity of cytotoxicity T lymphocytes. However, there is still a long way to go before the clinical application of D/M strategy. Although the efficiency of the strategy has been demonstrated in mice in this study, it needs further evaluation in advanced animal models (e.g., humanized mouse models) that are closer to human patients in terms of immune system, cancer cells, and tumor microenvironment. This study evaluated one designed D/M strategy. More D/M strategies, taking into account the variations in vaccine doses, immunization time points, and administration route (i.p., i.m., etc.), are needed to define the most effective strategy. Finally, we will explore the detailed anti-tumor mechanism of D/M immunization in future studies, including the importance of CD4+ and CD8+ T cells, the role of humoral immune response in the anti-tumor effects, and transcriptional changes in the immune cells in tumor microenvironment.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download ()Disclosure statement
The authors report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73. doi:10.1186/s12916-016-0623-5.
- Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20, 651–668.doi:10.1038/s41577-020-0306-5.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–12. doi:10.1038/nrc3239.
- He X, Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020. doi:10.1038/s41422-020-0343-4.
- Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642–662. doi:10.1038/nrd4663.
- Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14(8):559–567. doi:10.1038/nrc3770.
- Ledford H. Cancer-fighting viruses win approval. Nature. 2015;526(7575):622–623. doi:10.1038/526622a.
- Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, Wang XY. Therapeutic cancer vaccines: past, present, and future. Adv Cancer Res. 2013;119:421–475.
- van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer. 2016;16(4):219–233. doi:10.1038/nrc.2016.16.
- Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines. 2019;4:7. doi:10.1038/s41541-019-0103-y.
- Cheever MA, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. 2011;17(11):3520–3526. doi:10.1158/1078-0432.CCR-10-3126.
- Martinez-Saez N, Peregrina JM, Corzana F. Principles of mucin structure: implications for the rational design of cancer vaccines derived from MUC1-glycopeptides. Chem Soc Rev. 2017;46(23):7154–7175. doi:10.1039/C6CS00858E.
- Nath S, Mukherjee P. MUC1: a multifaceted oncoprotein with a key role in cancer progression. Trends Mol Med. 2014;20(6):332–342. doi:10.1016/j.molmed.2014.02.007.
- Beckwith DM, Cudic M. Tumor-associated O-glycans of MUC1: carriers of the glyco-code and targets for cancer vaccine design. Semin Immunol. 2020;47:101389. doi:10.1016/j.smim.2020.101389.
- Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8(1):61–70. doi:10.1038/nrc2293.
- Andersen MH, Pedersen LO, Capeller B, Brocker EB, Becker JC, thor Straten P. Spontaneous cytotoxic T-cell responses against survivin-derived MHC class I-restricted T-cell epitopes in situ as well as ex vivo in cancer patients. Cancer Res. 2001;61(16):5964–5968.
- Rohayem J, Diestelkoetter P, Weigle B, Oehmichen A, Schmitz M, Mehlhorn J et al. Antibody response to the tumor-associated inhibitor of apoptosis protein survivin in cancer patients. Cancer Res. 2000;60(7):1815–1817.
- Garg H, Suri P, Gupta JC, Talwar GP, Dubey S. Survivin: a unique target for tumor therapy. Cancer Cell Int. 2016;16:49. doi:10.1186/s12935-016-0326-1.
- Rapoport AP, Aqui NA, Stadtmauer EA, Vogl DT, Fang H-B, Cai L, Janofsky S, Chew A, Storek J, Akpek G, et al. Combination immunotherapy using adoptive T-cell transfer and tumor antigen vaccination on the basis of hTERT and survivin after ASCT for myeloma. Blood. 2011;117(3):788–797. doi:10.1182/blood-2010-08-299396.
- Gaidzik N, Westerlind U, Kunz H. The development of synthetic antitumour vaccines from mucin glycopeptide antigens. Chem Soc Rev. 2013;42(10):4421–4442. doi:10.1039/c3cs35470a.
- Acres B, Lacoste G, Limacher JM. Targeted Immunotherapy Designed to Treat MUC1-Expressing Solid Tumour. Curr Top Microbiol Immunol. 2017;405:79–97.
- Disis ML, Gad E, Herendeen DR, Lai V-P, Park KH, Cecil DL, O’Meara MM, Treuting PM, Lubet RA. A multiantigen vaccine targeting neu, IGFBP-2, and IGF-IR prevents tumor progression in mice with preinvasive breast disease. Cancer Prev Res (Phila). 2013;6(12):1273–1282. doi:10.1158/1940-6207.CAPR-13-0182.
- Chen J, Guo X-Z, Li H-Y, Liu X, Ren L-N, Wang D, Zhao -J-J. Generation of CTL responses against pancreatic cancer in vitro using dendritic cells co-transfected with MUC4 and survivin RNA. Vaccine. 2013;31(41):4585–4590. doi:10.1016/j.vaccine.2013.07.055.
- Gatti-Mays ME, Strauss J, Donahue RN, Palena C, Del Rivero J, Redman JM, Madan RA, Marté JL, Cordes LM, Lamping E, et al. A Phase I Dose-Escalation Trial of BN-CV301, a Recombinant Poxviral Vaccine Targeting MUC1 and CEA with Costimulatory Molecules. Clin Cancer Res. 2019;25(16):4933–4944. doi:10.1158/1078-0432.CCR-19-0183.
- Zhang H, Liu C, Zhang F, Geng F, Xia Q, Lu Z, Xu P, Xie Y, Wu H, Yu B, et al. MUC1 and survivin combination tumor gene vaccine generates specific immune responses and anti-tumor effects in a murine melanoma model. Vaccine. 2016;34(24):2648–2655. doi:10.1016/j.vaccine.2016.04.045.
- Liu C, Lu Z, Xie Y, Guo Q, Geng F, Sun B, Wu H, Yu B, Wu J, Zhang H, et al. Soluble PD-1-based vaccine targeting MUC1 VNTR and survivin improves anti-tumor effect. Immunol Lett. 2018;200:33–42. doi:10.1016/j.imlet.2018.06.004.
- Rice J, Ottensmeier CH, Stevenson FK. DNA vaccines: precision tools for activating effective immunity against cancer. Nat Rev Cancer. 2008;8(2):108–120. doi:10.1038/nrc2326.
- Grunwald T, Tenbusch M, Schulte R, Raue K, Wolf H, Hannaman D, de Swart RL, Uberla K, Stahl-Hennig C. Novel vaccine regimen elicits strong airway immune responses and control of respiratory syncytial virus in nonhuman primates. J Virol. 2014;88(8):3997–4007. doi:10.1128/JVI.02736-13.
- Gary EN, Weiner DB. DNA vaccines: prime time is now. Curr Opin Immunol. 2020;65:21–27. doi:10.1016/j.coi.2020.01.006.
- Lim M, Badruddoza AZM, Firdous J, Azad M, Mannan A, Al-Hilal TA, Cho C-S, Islam MA. Engineered Nanodelivery Systems to Improve DNA Vaccine Technologies. Pharmaceutics. 2020;12(1). doi:10.3390/pharmaceutics12010030.
- Gomez-Aguado I, Rodriguez-Castejon J, Vicente-Pascual M, Rodriguez-Gascon A, Solinis MA, Del Pozo-Rodriguez A. Nanomedicines to deliver mRNA: state of the art and future perspectives. Nanomaterials (Basel). 2020;10(2):364.doi:10.3390/nano10020364.
- DeMuth PC, Min Y, Huang B, Kramer JA, Miller AD, Barouch DH, Hammond PT, Irvine DJ. Polymer multilayer tattooing for enhanced DNA vaccination. Nat Mater. 2013;12(4):367–376. doi:10.1038/nmat3550.
- Reparaz D, Llopiz D, Sarobe P. When Cancer Vaccines Go Viral. Clin Cancer Res. 2019;25(16):4871–4873. doi:10.1158/1078-0432.CCR-19-1652.
- Cottingham MG, Carroll MW. Recombinant MVA vaccines: dispelling the myths. Vaccine. 2013;31(39):4247–4251. doi:10.1016/j.vaccine.2013.03.021.
- Lu S. Heterologous prime-boost vaccination. Curr Opin Immunol. 2009;21(3):346–351. doi:10.1016/j.coi.2009.05.016.
- McConkey SJ, Reece WHH, Moorthy VS, Webster D, Dunachie S, Butcher G, Vuola JM, Blanchard TJ, Gothard P, Watkins K, et al. Enhanced T-cell immunogenicity of plasmid DNA vaccines boosted by recombinant modified vaccinia virus Ankara in humans. Nat Med. 2003;9(6):729–735. doi:10.1038/nm881.
- Brown SA, Surman SL, Sealy R, Jones BG, Slobod KS, Branum K, Lockey TD, Howlett N, Freiden P, Flynn P, et al. Heterologous Prime-Boost HIV-1 Vaccination Regimens in Pre-Clinical and Clinical Trials. Viruses. 2010;2(2):435–467. doi:10.3390/v2020435.
- Hallengard D, Lum F-M, Kummerer BM, Lulla A, Lulla V, Garcia-Arriaza J, Fazakerley JK, Roques P, Le Grand R, Merits A, et al. Prime-boost immunization strategies against Chikungunya virus. J Virol. 2014;88(22):13333–13343. doi:10.1128/JVI.01926-14.
- Smith CL, Dunbar PR, Mirza F, Palmowski MJ, Shepherd D, Gilbert SC, Coulie P, Schneider J, Hoffman E, Hawkins R, et al. Recombinant modified vaccinia Ankara primes functionally activated CTL specific for a melanoma tumor antigen epitope in melanoma patients with a high risk of disease recurrence. Int J Cancer. 2005;113(2):259–266. doi:10.1002/ijc.20569.
- Kim SB, Ahn J-H, Kim J, Jung KH. A phase 1 study of a heterologous prime-boost vaccination involving a truncated HER2 sequence in patients with HER2-expressing breast cancer. Mol Ther Methods Clin Dev. 2015;2:15031. doi:10.1038/mtm.2015.31.
- Zhang H, Wang Y, Liu C, Zhang L, Xia Q, Zhang Y, Wu J, Jiang C, Chen Y, Wu Y, et al. DNA and adenovirus tumor vaccine expressing truncated survivin generates specific immune responses and anti-tumor effects in a murine melanoma model. Cancer Immunol Immunother. 2012;61(10):1857–1867. doi:10.1007/s00262-012-1296-3.
- Wang YQ, Zhang -H-H, Liu C-L, Wu H, Wang P, Xia Q, Zhang L-X, Li B, Wu J-X, Yu B, et al. Enhancement of survivin-specific anti-tumor immunity by adenovirus prime protein-boost immunity strategy with DDA/MPL adjuvant in a murine melanoma model. Int Immunopharmacol. 2013;17(1):9–17. doi:10.1016/j.intimp.2013.04.015.
- Xu R, Johnson AJ, Liggitt D, Bevan MJ. Cellular and humoral immunity against vaccinia virus infection of mice. J Immunol. 2004;172(10):6265–6271. doi:10.4049/jimmunol.172.10.6265.
- Park A, Fong Y, Kim S, Yang J, Murad J, Lu J et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci Transl Med. 2020;12(559)eaaz1863. doi:10.1126/scitranslmed.aaz1863.
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–221. doi:10.1038/nature22991.
- Hammerich L, Marron TU, Upadhyay R, Svensson-Arvelund J, Dhainaut M, Hussein S et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat Med. 2019;25(5):814–824. doi:10.1038/s41591-019-0410-x.
- Shibata T, Lieblong BJ, Sasagawa T, Nakagawa M. The promise of combining cancer vaccine and checkpoint blockade for treating HPV-related cancer. Cancer Treat Rev. 2019;78:8–16. doi:10.1016/j.ctrv.2019.07.001.
- Geng F, Bao X, Dong L, Guo -Q-Q, Guo J, Xie Y, Zhou Y, Yu B, Wu H, Wu J-X, et al. Doxorubicin pretreatment enhances FAPalpha/survivin co-targeting DNA vaccine anti-tumor activity primarily through decreasing peripheral MDSCs in the 4T1 murine breast cancer model. Oncoimmunology. 2020;9(1):1747350. doi:10.1080/2162402X.2020.1747350.
- Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci U S A. 2015;112(6):1809–1814. doi:10.1073/pnas.1417636112.
- Hillman GG, Reich LA, Rothstein SE, Abernathy LM, Fountain MD, Hankerd K, Yunker CK, Rakowski JT, Quemeneur E, Slos P, et al. Radiotherapy and MVA-MUC1-IL-2 vaccine act synergistically for inducing specific immunity to MUC-1 tumor antigen. J Immunother Cancer. 2017;5:4. doi:10.1186/s40425-016-0204-3.
- Demaria S, Golden EB, Formenti SC. Role of local radiation therapy in cancer immunotherapy. JAMA Oncol. 2015;1(9):1325–1332. doi:10.1001/jamaoncol.2015.2756.