
ABSTRACT
CD8+ T cells are capable of recognizing mutation-derived neoantigens displayed by HLA class I molecules, thereby exhibiting the ability to distinguish between cancer and normal cells. However, accumulating evidence has shown that only a small fraction of nonsynonymous somatic mutations give rise to clinically relevant neoantigens. The properties of such neoantigens, which must be presented by HLA and immunogenic to induce a T-cell response, remain elusive. In this study, we explored the HLA class I ligandome of a human cancer cell line with microsatellite instability using a proteogenomic approach. The results demonstrated that neoantigens accounted for only 0.34% of the HLA class I ligandome, and most neoantigens were encoded by genes with abundant expression. Thereafter, T-cell responses were prioritized, and immunodominant neoantigens were defined using naive CD8+ T cells derived from healthy donors. AKF9, an immunogenic neoantigen with a mutation at a non-anchor position, formed a stable peptide-HLA complex. T-cell responses were analyzed against a panel of AKF9 variants with single amino-acid substitutions, in which mutations did not alter the high HLA-binding affinity and stability. The responses varied across individuals, demonstrating the impact of heterogeneous T-cell repertoires in this human cancer model. Moreover, responses were biased toward a variant group with large structural changes compared to the wild-type peptide. Thus, naive T-cell induction can be attributed to multiple determinants. Combining structural dissimilarity with gene-expression levels, HLA-binding affinity, and stability may further help prioritize the immunogenicity of non-anchor-type neoantigens.
Introduction
Nonsynonymous somatic mutations that occur in the cancer genome give rise to mutated HLA ligands, which are referred to as neoantigens. Because neoantigens are absent in normal tissues, they are not tolerated by the host immune system, thereby eliciting an anti-cancer response.Citation1 Clinical evidence supports the pivotal role of neoantigens in engaging cytotoxic CD8+ T cells in immune surveillance against cancer. Mismatch repair deficiency (dMMR), or microsatellite instability (MSI), serves as a tissue agnostic biomarker predictive of favorable prognosis in melanoma and solid tumor patients treated with immune checkpoint blockade.Citation2–6 Moreover, T-cell subsets recognizing neoantigens are often found in tumor-infiltrating lymphocytes (TILs), and the adoptive transfer of these neoantigen-reactive T cells leads to tumor regression in a variety of tumor types.Citation7–11 Thus, the cancer-specific host T-cell response elicited by neoantigens is capable of controlling tumor growth and holds great promise for the development of cancer immunotherapy. Meanwhile, most somatic mutations are random events that occur in the cancer genome, and neoantigens are therefore unique to individuals. Furthermore, the spontaneous host T-cell response is limited to only a small fraction of nonsynonymous somatic mutations,Citation9,Citation12 indicating the rarity of clinically relevant neoantigens. Although neoantigens must be identified per patient for clinical interventions, the molecular properties, or determinants that differentiate a limited number of clinically relevant neoantigens from non-immunogenic mutated peptides, are not fully understood, leaving the specificity of in silico prediction unsatisfactory.
Clinically relevant neoantigens must be naturally presented by HLA and immunogenic to induce host T-cell responses. Recent technological advances in detecting HLA-bound peptides using mass spectrometry (MS) have enabled the direct and comprehensive analysis of thousands of natural HLA ligands, including neoantigens.Citation13–21 In sharp contrast to approaches using prediction algorithms based solely on HLA-binding affinity, the use of MS offers an unbiased way to analyze part of, if not all, the landscape of the truly presented immunopeptidome as a whole.Citation22–24 These natural HLA ligand data have contributed to the detection of antigen processing signatures and the development of prediction algorithms.Citation25–27 Meanwhile, in silico-predicted HLA-binding affinity may prioritize the immunogenicity of neoantigens.Citation28 A seminal study has proposed the differential HLA-binding affinity between neoantigens and wild-type peptides (WTs) as a predictor of immunogenicity, because WTs with low binding affinity are not presented, thereby making neoantigens new to host immune surveillance.Citation29 A recent study has also shown the contribution of the absolute values of binding affinity in the prediction of immunogenic non-anchor-type neoantigens that harbor mutations outside of the HLA-binding anchor positions.Citation30
In the present study, an MSI-high human colorectal cancer (CRC) model with high tumor mutation burden (TMB) was examined, and a non-anchor-type neoantigen, AKF9, was identified, which predominantly induced CD8+ T-cell responses among naturally presented neoantigens. Further analysis using a panel of single amino-acid substitutions with comparable HLA binding affinity demonstrated a hierarchy of neoantigen variants in T-cell induction. The determinants of immunogenicity among non-anchor-type neoantigens with comparable HLA binding affinities are not clear. This experimental model demonstrated that structural differences between neoantigens and WTs were, in part, correlated with their immunogenicity.
Results
Discovery of neoantigens that were naturally presented by MSI-CRC cells
An MSI-CRC cell line (HCT15) was used as an experimental model because of its prevalent HLA class I genotypes (HLA-A*02:01 and HLA-A*24:02) and high TMB, which yields nonsynonymous mutations.Citation31 In addition, HCT15 may retain unedited immunogenic neoantigens because of the mutated beta-2-microglobulin (B2M) gene, leading to a loss of HLA presentation.Citation32 Therefore, the B2M gene was reconstituted and a stably expressed HCT15/β2 m cell line was prepared (Figure S1).Citation17,Citation33 To evaluate the natural HLA class I ligandome and neoantigens, HLA class I ligands were immunoprecipitated using a pan-HLA class I monoclonal antibody and the eluted ligands were analyzed using tandem mass spectrometry (MS/MS). The obtained MS/MS spectra were used to search a custom reference database containing virtually translated polypeptides that arise from somatic 2,241 missense and 32 frameshift mutations, as well as known protein sequences. Proteogenomic analysis identified the landscape of the HLA class I repertoire comprising 2,352 non-redundant HLA class I ligands (Table S1), including eight unique neoantigens at a stringent false discovery rate (FDR) of 0.01 (, , Figure S2 and S3). Prediction using NetMHCpan-4.1 assigned the peptides across HLA class I types responsible for presentation. Indeed, the number of identified peptides varied across HLA types and was positively correlated with the surface expression levels of the corresponding HLA molecules (Figure S1). Although the possibility of potential neoantigens left undetected in this proteogenomic approach was not ruled out and the analysis was restricted to exonic mutations, the results showed that natural neoantigens accounted for a small and limited subset (0.34%, 8 out of 2,352) of an HLA ligandome in this MSI-CRC model with high TMB. In accordance with previous findings,Citation25,Citation34 natural HLA class I ligands, including neoantigens, originated from genes with abundant expression, indicating that gene expression levels influenced HLA class I presentation status (). The average gene expression of all HLA ligands and neoantigens was 153.9 and 166.6 RPKM, respectively, while that of the whole transcriptome was 53.1 RPKM (***p < .001).
Figure 1. Identification of HLA class I neoantigens that are naturally presented by microsatellite instability-colorectal cancer (MSI-CRC) cells. (A) Left, a pie chart showing the proportion of neoantigens in the entire HLA class I peptide repertoire detected by proteogenomic HLA ligandome analysis using mass spectrometry in HCT15/β2 m cells. Numbers in parentheses indicate the number of non-redundant peptides. Right, isolated HLA class I ligands were classified according to HLA type based on NetMHCpan-4.1%Rank scores. Each red dot represents detected neoantigens assigned to the indicated HLA types. (B) Differential gene expression between the whole transcriptome and source genes encoding HLA ligands in the transcriptome (*** p<.001, two-tailed t-test). Each red dot represents the distribution of source genes encoding neoantigens
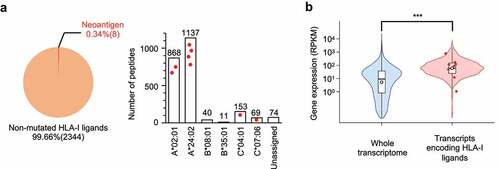
Table 1. Sequences and properties of neoantigens that are naturally presented by HLA class I of HCT15/β2m cells
CD8+ T-cell responses to naturally presented HLA-A24 neoantigens
The immunogenicity of antigens is defined as the ability to elicit a host immune response, which is influenced by both the host immune cell repertoire and intrinsic properties of antigens. To assess the immunogenicity of the detected neoantigens, peripheral blood derived mononuclear cells (PBMCs) were obtained from a panel of six healthy donors (HD) who did not have responsible gene mutations (Figure S4). Although the evaluation of patient-derived materials, such as TILs, directly shows the presence of spontaneous T-cell responses in cancer lesions, HD-derived T cells are potentially unbiased by the immunosuppressive tumor microenvironment; therefore, they often contain a broader repertoire of neoantigen-reactive T cells.Citation35 As such, a naive T-cell subset of HD PBMCs was cultured with autologous dendritic cells (DCs) pulsed with a cocktail of HLA-A24 neoantigens (Figure S5A and B).Citation36 The frequency of CD8+ T cells recognizing four natural neoantigens presented by HLA-A*24:02 was assessed using neoantigen-HLA-A24 tetramers before and after in vitro stimulation. Neoantigens with an increased frequency of responding T cells (≥ 4-fold increase compared with the naive population, with ≥ 0.1% of tetramer+ CD8+ cells) were considered immunogenic in this setting. Here, varied responses across six HD PBMCs were observed, where four HDs responded to one or more neoantigens, while the other two HDs did not respond, indicating heterogeneity among individuals in the host T-cell repertoire responding to the neoantigen (, B, and Figure S6A.
Figure 2. Immunogenicity of HLA class I neoantigens inducing healthy donor (HD)-naive CD8+ T-cell responses. Naive CD8+ T cells derived from HD-PBMCs were stimulated with autologous dendritic cells (DCs) pulsed with a cocktail of HLA-A24 neoantigens along with CMVpp65. On days −1 (before stimulation) and 11 (after stimulation), the frequency of reactive CD8+ T cells was measured using the corresponding peptide-HLA-A24 tetramers. (A) Representative responses of T-cell induction in HD5 cells. Numbers indicate the proportion of tetramer-positive CD8+ cells. (B) Summary of T-cell induction in six HD-PBMCs. The frequency of CD8+ T cells recognizing four unique HLA-A24 neoantigens before and after stimulation is shown. Asterisks indicate T-cell responses in which the frequency increased ≥ 4-fold after stimulation, with ≥ 0.1% tetramer+ CD8+ cells. (C) Peptide-HLA-A24 stability assay using T2-A24 cells in a range of indicated peptide concentrations. The change in mean fluorescence intensity (ΔMFI) was calculated as the experimental MFI minus the background MFI without a peptide pulse. The stability of neoantigens (red) and WT (blue) is shown. CMVp65 and GK12 served as positive and negative HLA-A24-binding controls, respectively. (D) Comparison of ∆MFI in a peptide-HLA class I stability assay at a peptide concentration of 1 µM. Error bars represent SEM (n = 3). *p<.05, **p<.01, ns, not significant, two-tailed t-test
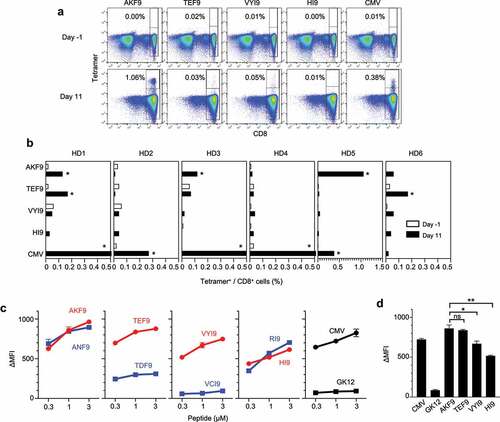
Meanwhile, a trend in the immunogenicity of these neoantigens was observed. AYLEAIHKF (AKF9) and TLPEEFHEF (TEF9) were found to be immunogenic, eliciting CD8+ T-cell responses in three and two HDs, respectively. We previously reported the cytotoxicity and specificity of CD8+ T cells induced by AKF9.Citation17 Both neoantigens arose from missense mutations in genes with abundant expression (AP2S1 and RAD21), harboring amino-acid substitutions at position 8 (P8). The p.N86K mutation in AP2S1 that gave rise to AKF9 did not influence HLA-A*24:02 binding because: (1) both the NetMHCpan-4.1 score and peptide-HLA class I (pHLA I) stability were unaltered by amino-acid substitutions, and (2) the WT was detected by HLA ligandome analysis ( and ). In contrast, the p.D116E mutation of TEF9 increased both the binding affinity and stability of pHLA I, suggesting that the substitution led to HLA presentation. Therefore, AKF9 and TEF9 were regarded as HLA-binding non-anchor and anchor-type neoantigens, respectively. Both types of neoantigens were capable of eliciting T-cell responses. Some possible properties that differentiated these two immunogenic neoantigens from other neoantigens included pHLA I stability and their HLA-binding affinity ().Citation35,Citation37 Regardless of the HLA-binding anchor type, both AKF9 and TEF9 showed significantly higher pHLA I stability, which was measured based on stable pHLA I formation on transporter associated with antigen processing (TAP)-deficient T2-A24 cells, than the other two natural neoantigens (VYI9 and HI9) that failed to induce T-cell responses. Thus, the immunogenicity of the HLA-A24 neoantigens was prioritized, whereby AKF9 predominantly induced CD8+ T-cell responses in three of six HD-PBMCs.
A panel of AKF9 variants with single amino-acid substitutions
Given the absence of the wild-type presentation of TEF9, it is plausible that an abrupt HLA presentation of TEF9 was new to the HD T-cell repertoires, eliciting a T-cell response. In accordance with this notion, the difference in MHC binding affinity between neoantigens and WTs is correlated with the immunogenicity observed both in preclinical mouse models and human samples.Citation29,Citation38–40 Meanwhile, many nonsynonymous somatic mutations potentially give rise to non-anchor-type neoantigens, in which mutations do not affect HLA presentation. Capietto et al. proposed the absolute values of HLA-binding affinity as a predictor of immunogenicity of non-anchor-type neoantigens.Citation30 However, non-anchor-type neoantigens with high binding affinity do not always induce T-cell responses, and the intrinsic properties of neoantigens that prioritize immunogenicity remain unclear. Because WTs were simultaneously presented by the HLA in this setting, T cells must sense and discriminate changes in amino-acid substitutions.
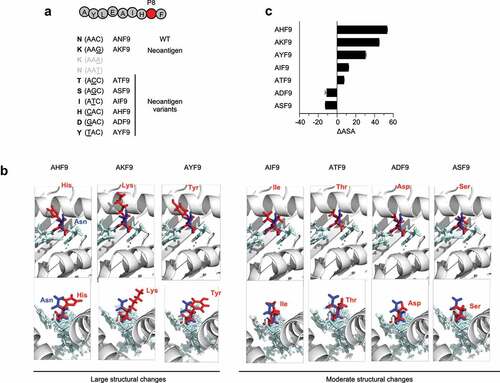
Here, we hypothesized that structural changes in mutated residues confer immunogenicity to non-anchor-type neoantigens. To test this hypothesis, AKF9 was chosen as a model, and its single amino-acid substitution variants were prepared based on single-nucleotide missense mutations that possibly occurred within the same codon (). Although P8 is next to the C-terminal residue that projects toward the deep F-pocket of HLA class I in a 9-mer peptide, P8 is often exposed to TCR contact surfaces.Citation41 3D modeling of the complexes of the AKF9 variants and HLA-A*24:02 classified variants into two groups: a variant group with large structural changes, where the positively charged or aromatic side chains of His, Lys, and Tyr protruded toward the solvent surface faced with a T-cell receptor (TCR), and another variant group with moderate structural changes caused by the substitution of Ile, Thr, Asp, and Ser for the wild-type peptide, Asn (). To validate the structural differences, accessible surface areas (ASA) of neoantigen peptides predicted by the online server PEP-FOLD3.5 were employed ().Citation42 While this approach did not take into account the influence of an HLA molecule forming a pHLA I complex, it allowed the assessment of the surface areas of neoantigens without using crystal structure data. As anticipated, the difference in ASA between each AKF9 variant and the WT (∆ASA) demonstrated a difference between the group with large structural changes (mean ∆ASA: 43.0) and those with moderate changes (mean ∆ASA: −0.9).
Figure 3. 3D-Modeling of AKF9 variants with single amino-acid substitutions complexed with HLA-A24. (A) Variants of the immunogenic AKF9 neoantigen, where wild-type Asn at position 8 (P8) was substituted with the indicated amino acids encoded by possible single nucleotide mutations within the wild-type codon (AAC). (B) 3D-modeling of the neoantigen-HLA-A24 complexes. Neoantigen complexes superimposed on the wild-type complex are shown. The upper and lower panels show the view from the top (T-cell contact surface) and the side (C-terminal end of the peptide) of the complexes, respectively. The ribbon diagram in gray represents the α1 and α2 helices of an HLA-A24 molecule. Stick models in light blue represent a peptide backbone with the side chains of mutated (red) and wild-type (blue) residues at P8. (C) Differences in the accessible surface area between the neoantigen and the WT (∆ASA) of AKF9 variants. Error bars represent the SEM (n = 3)
Immunogenic prioritization of AKF9 variants with structural differences
Any substitutions were stably presented by HLA-A*24:02, where neither HLA binding affinity nor stability stratified the variants ( and ). However, the immunogenicity to elicit T-cell responses in the seven HD PBMCs was not comparable. Stimulation using autologous DCs pulsed with a cocktail of the AKF9 variants along with the WT demonstrated that substitutions from Asn to His, Lys, Tyr, and Asp (in AHF9, AKF9, AYF9, and ADF9, respectively) increased the frequency of reactive CD8+ T cells, as assessed by staining with neoantigen-HLA-A24 tetramers, while other variants, including the WT, failed to elicit responses (, C and Figure S6B). Although we cannot exclude the possibility that T-cell precursors responding to ‘non-immunogenic’ neoantigens were not included due to the limited number of naive T cells in the panel of HD-derived samples, the results showed that T-cell responses varied, even among mutations that did not alter HLA binding affinity and stability. In this experimental setting, the variants with large structural changes (3 of 3), in contrast to the variants with moderate structural changes (1 of 4), predominantly induced CD8+ T-cell responses.
Figure 4. Immunogenic prioritization of non-anchor-type neoantigen variants with single amino-acid substitutions. (A) Peptide-HLA-A24 stability assay using T2-A24 cells in a range of indicated peptide concentrations. The change in mean fluorescence intensity (ΔMFI) was calculated as the experimental MFI minus the background MFI without a peptide pulse. (B) Representative responses of T-cell induction against AKF9 and ADF9. Numbers indicate the proportion of tetramer-positive CD8+ cells. Naive CD8+ T cells derived from HD-PBMCs were stimulated with autologous DCs pulsed with a cocktail of neoantigen variants along with the WT and CMVpp65. On days −1 (before stimulation) and 11 (after stimulation), the frequency of reactive CD8+ T cells was measured using the corresponding peptide-HLA-A24 tetramers. (C) Summary of T-cell induction in seven HD-PBMCs. The frequency of CD8+ T cells recognizing neoantigen variants or the WT before and after stimulation is shown. Asterisks indicate T-cell responses in which the frequency increased ≥ 4-fold after stimulation, with ≥ 0.1% tetramer+ CD8+ cells. The HDs with the same numbers are in common with those in
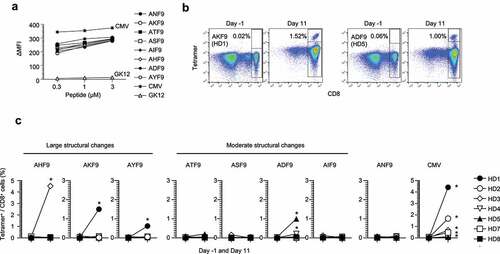
Table 2. Physicochemical properties of AKF9 neoantigen variants
Despite a previous notion of the preference for hydrophobic TCR-contact residues in immunogenic neoantigens,Citation43,Citation44 hydrophobicity was not a determinant in this model. An alternative physicochemical property possibly linked to immunogenicity was the difference in charge status between neoantigens and the WT (). Therefore, the structural difference, possibly along with altered charge status, may allow TCRs to discriminate non-anchor-type neoantigens from the wild-type counterpart.
Functions, specificity, and cross reactivity of neoantigen-reactive T cells
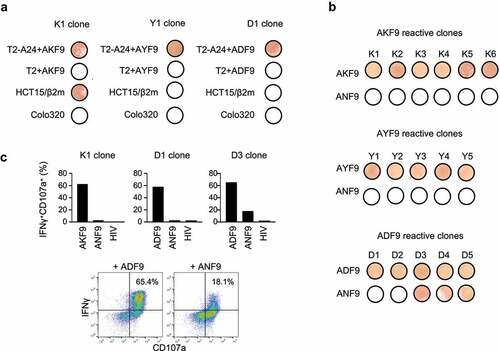
Lastly, to assess induced T-cell functions, CD8+ subsets positive for neoantigen-HLA-A24 tetramers were sorted, and multiple CD8+ T-cell clones were established for each of the three immunogenic AKF9 variants. The clones reactive to AKF9 responded to AKF9 only in the presence of HLA-A24, and produced IFNγ in response to HCT15/β2 m cells that had the corresponding AP2S1 mutation. This demonstrated that the T-cell response functions in an HLA-A24 restricted manner as well as the natural presentation of the AKF9 neoantigen (). Likewise, both AYF9 and ADF9 variants induced reactive clones; however, the clones failed to recognize HCT15/β2m cells as expected. Thus, this approach using naive CD8+ T cells, along with autologous DCs, induced functional CD8+ T cells, and induction rates of T-cell subsets positive for tetramer staining represented the immunogenicity of the corresponding neoantigens. In addition, while the AKF9 and AYF9-clones ignored the WT, three of the five clones reactive to ADF9 simultaneously recognized ANF9 (). It is well known that some CD8+ T cell subsets, including neoantigen-reactive T cells, potentially cross-react with other peptides or WTs, most likely to compensate for a limited number of TCR repertoires.Citation45–47 The mutation-specific response of an AKF9-reactive clone was also confirmed by intracellular IFNγ production and surface CD107a expression measured by flow cytometry (). The D1 clone also showed a specific response to ADF9. In contrast, the D3 clone responded not only to ADF9 but also to ANF9, supporting the cross-reactivity of the T-cell clone. Our observations indicated that ADF9, with a moderate structural change, induced at least two different types of CD8+ clones with functionally distinct TCRs, one of which failed to discriminate the neoantigen from the WT.
Figure 5. Functions and specificity of induced CD8+ T cells reactive to neoantigen variants. (A) IFNγ ELISpot assay of representative AKF9 (K1), AYF9 (Y1), and ADF9 (D1)-reactive CD8+ T-cell clones against T2-A24 or T2 pulsed with 20 µM indicated peptides, or cancer cells without a peptide pulse. (B) Cross-reactivity of neoantigen-reactive CD8+ T-cell clones. IFNγ ELISpot assay of established clones reactive to AKF9 (K1-K6), AYF9 (Y1-Y5), and ADF9 (D1-D5) is shown against T2-A24 pulsed with the indicated neoantigens, or the WT. (C) Intracellular IFNγ production and CD107a surface expression of neoantigen-reactive T-cell clones measured using flow cytometry. In bar graphs, the responses of the indicated clones against T2-A24 pulsed with 20 µM indicated peptides are summarized. The y-axis shows the proportions of IFNγ and CD107a double-positive cells. In the density plots, representative responses of D3 are shown. The data are representative of three independent experiments
Discussion
Among nonsynonymous somatic mutations that occur in cancer, only a limited number of mutations are ultimately presented by HLA and give rise to clinically relevant neoantigens capable of inducing a host T-cell response. In the present study, the HLA class I repertoire of a human MSI-CRC model was investigated, and properties of neoantigens required for immunogenicity and HLA presentation were assessed. A proteogenomic approach enabled the direct and comprehensive detection of neoantigens along with non-mutated HLA class I ligands, showing the rarity of naturally presented neoantigens even in the mutation-rich CRC model. The neoantigens originated from genes with abundant expression as is the case with non-mutated HLA class I ligands.Citation25,Citation34 The HLA class I ligandome contained both anchor and non-anchor-type neoantigens, and we found that a non-anchor-type neoantigen, AKF9, predominantly induced T-cell response. Because cells constantly display thousands of self-peptides by HLA, which are tolerated by host T cells, the immunogenicity of neoantigens could be attributed to dissimilarity to self-peptides. For instance, frameshifts caused by indel mutations may yield more immunogenic neoantigens, as their WTs are, in principle, absent; therefore, every neoantigen becomes new to the host.Citation48,Citation49 Even among missense mutations, amino-acid substitutions at HLA-binding anchor positions may follow the same principle.Citation29 In contrast, non-anchor-type neoantigens, which are supposed to be dominant in numbers, also induce CD8+ T-cell responses. In this case, HLA-binding affinity serves as a predictor of immunogenicity, as shown by preclinical mouse models.Citation30 Here, non-anchor-type neoantigens were further dissected, demonstrating that, even among those with comparable binding affinity or stability, there were differences in immunogenicity, which could be prioritized by their structural differences. Increased ∆ASA in immunogenic neoantigens may imply that peptide volume gains by mutations cause structural changes that are detected by T cells. These findings underscore the necessity of classification according to neoantigen type in assessing immunogenicity.
While neoantigens are believed to be specific to cancer, reactive T cells often cross-react with WTs.Citation30,Citation46 Although we observed a trend in eliciting T-cell responses by mutated residues with structural changes, our results do not underestimate the potential of other possible determinants. Among the AKF9 variants, ADF9, with a moderate structural change, induced reactive CD8+ T cells. The determinant of immunogenicity in this case remained elusive, possibly due to a relatively large change in the net charge values caused by the substitution. Interestingly, however, only the ADF9 variant induced reactive CD8+ T cells that cross-reacted with the WT. Indeed, the substitution of Asp for Asn shows the highest PAM1 value among possible substitutions originating in Asn, which is consistent with a previously identified positive correlation between PAM1 values and cross-reactivity.Citation46 Currently, it is not clear whether the cross-reactivity caused by neoantigen-reactive T cells leads to the development of autoimmune diseases. Induction of T cells reactive to neoantigens with moderate structural changes could lead to cross-reactivity to WTs; however, accumulation of clinical evidence regarding cross-reactive T cells, including studies of cells induced by vaccination, would be required to validate this hypothesis.
A technical limitation in the structural assessment is the lack of accuracy in scoring the precise structure of pHLA I, in particular, the differences between neoantigens and WTs bound to HLA molecules. Structural simulation of a neoantigen-HLA I complex often exploits an established crystallographic structure as a template, optimizing the complex with a replaced peptide sequence of interest.Citation13 This method enables the specific measurement of the area of the solvent surface that T cells have contact with. However, the number of HLA types available for standards is limited; therefore, a broad assessment of neoantigens with different peptide lengths presented by a variety of HLA alleles is challenging.Citation41 Although ∆ASA can be applied to any peptide without established structural data of HLA types of interest, methodological improvement in the in silico simulation in a realistic setting would further amend this issue and contribute to precise immunogenicity prediction.
In addition to peptide-intrinsic determinants, interindividual heterogeneity in responsiveness to each natural neoantigen and AKF9 neoantigen model has been observed. It is important to note the possibility that a limited number of naive T cells included in a sample could not represent the entire T-cell repertoire present in the corresponding individual, resulting in the absence of responses in our experimental setting. Meanwhile, heterogeneity suggests a diversity of T-cell repertoires and other host-side factors. For instance, different infection histories or intestinal microbiota may sculpt the T-cell repertoire of an individual. Generated microbial-specific memory T cells can cross-react with neoantigens, and consequently lead to heterogeneous responses among different individuals.Citation50,Citation51 Indeed, T-cell cross-reactivity against microbial antigens and neoantigens has been observed in long-term survivors of pancreatic cancer.Citation52 Such TCR recognition probability derived from homology to microbial antigens can be estimated and used to improve immunogenic neoantigen prediction.Citation38,Citation53 Thus, not only peptide-intrinsic factors but also host-specific factors must be taken into account when determining the immunogenicity of neoantigens in clinical settings.
In conclusion, the HLA class I ligandome of human MSI-CRC cells was investigated and immunogenic neoantigens were identified. Structural differences between non-anchor-type neoantigens and WTs prioritized the immunogenicity of the model neoantigens. As shown in the case of ADF9, we showed that T cells are still able to recognize non-anchor-type neoantigens with moderate differences. In addition, our data are based on a limited number of HD samples and neoantigen models focusing on variations at P8. Therefore, further assessments using a variety of non-anchor-type neoantigens in a large scale are indispensable. Nevertheless, our current findings underscore the need for stratification according to the types of neoantigens, and may help predict immunogenic neoantigens from a vast array of somatic mutations and develop effective and specific clinical interventions targeting neoantigens.
Methods
Cells and culture
CRC cell lines HCT15 and Colo320 were purchased from ATCC, and HCT15/β2m cells that stably expressed the intact beta-2-microglobulin gene were established.Citation33 Unless specifically mentioned, cells were cultured in complete RPMI1640 or DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, 1% GlutaMAX (Gibco), 10 mM HEPES, 1 mM sodium pyruvate, and 55 µM 2-mercaptoethanol in a 5% CO2 incubator at 37°C. The T2-A24 cell line (T2 stably expressing HLA-A*24:02) was a gift from Dr. K. Kuzushima (Aichi Cancer Center Research Institute). The HLA class I genotype of HCT15 is as follows: A*24:02, A*02:01, B*08:01, B*35:01, C*04:01, and C*07:06. For gene expression and HLA genotype profiling, the data deposited in the TRON Cell Line Portal (http://celllines.tron-mainz.de) were used.Citation54
HD-derived PBMCs
The study was performed with the approval of the Research Ethics Committee of Sapporo Medical University (29-2-69). All HDs were confirmed to be HLA-A*24:02 positive by PCR of genomic DNA.Citation55 Peripheral blood was used to isolate PBMCs using Lymphoprep (Cosmo Bio), according to the manufacturer’s instructions. PBMCs were cultured in complete AIM-V medium (Gibco) containing 1% penicillin-streptomycin, 1% GlutaMAX (Gibco), 10 mM HEPES, 1 mM sodium pyruvate, 55 µM 2-mercaptoethanol, and 10% human AB serum.
Isolation of HLA-class I ligands
An established protocol was followed with slight modifications.Citation26,Citation56 A frozen cell pellet of 1.0 × 109 HCT15/β2 m cells was lysed with buffer containing 0.25% sodium deoxycholate, 0.2 mM iodoacetamide, 1 mM EDTA, protease inhibitor cocktail (Sigma), 1 mM PMSF, and 1% octyl-β-D glucopyranoside (Dojindo). The peptide-HLA class I complexes were captured by affinity chromatography using a purified pan-HLA class I monoclonal antibody (W6/32, ATCC) coupled to CNBr-activated Sepharose 4B (GE Healthcare). The peptides bound to HLA class I were eluted with mild acid (0.2% TFA) and desalted using a Sep-Pak tC18 cartridge (Waters) with 28% ACN in 0.1% TFA and ZipTip U-C18 (Millipore) with 50% ACN in 1% FA. Samples were dried using vacuum centrifugation and resuspended in 5% ACN in 0.1% TFA for LC-MS/MS analysis.
MS data acquisition
Samples were loaded into a nano-flow LC (Easy-nLC 1000 system, Thermo) online-coupled to an Orbitrap mass spectrometer equipped with a nanospray ion source (Q Exactive Plus, Thermo). In MS data acquisition, survey scan spectra were acquired at a resolution of 70,000 at 200 m/z with an AGC target value of 3e6 ions and a maximum IT of 100 ms, ranging from 350 to 2,000 m/z with charge states between 1+ and 4 + . A data-dependent top 10 method was employed. The MS/MS resolution was 17,500 at 200 m/z with an AGC target value of 1e5 ions and a maximum IT of 120 ms.
MS data analysis and neoantigen identification
To detect neoantigens, custom MS reference databases specific to the sample were built. HCT15 somatic missense and frameshift mutations were obtained from the COSMIC database (https://cancer.sanger.ac.uk/cosmic).Citation57 The nonsynonymous mutations unique to HCT15 were translated in frame and the polypeptide sequences (61-mer amino acids each) encompassing mutations were integrated into the conventional protein reference database (Swiss-Prot). Next, MS/MS data were used to search the custom databases using Sequest HT on the Proteome Discoverer platform (Thermo) with tolerance of precursor and fragment ions of 10 ppm and 0.02 Da, respectively. No specific enzyme was selected for the search, and the oxidation of methionine was selected as a dynamic modification. A stringent false discovery rate (FDR) of 0.01 was used in the Percolator node of the Proteome Discoverer version 2.3 software (Thermo) as a peptide detection threshold. The HLA class I types of detected neoantigens and non-mutated ligands were determined according to their NetMHCpan-4.1 scores. The sequences with %Rank scores of 2.0 or more against any of the HCT15 HLA class I alleles were left unassigned (Table S1).
Synthetic peptides and HLA tetramers
Synthetic peptides (GYISPYFINTSK, GK12; QYDPVAALF, CMVpp65; AYLEAIHNF, ANF9; AYLEAIHKF, AKF9; AYLEAIHTF, ATF9; AYLEAIHSF, ASF9; AYLEAIHIF, AIF9; AYLEAIHHF, AHF9; AYLEAIHDF, ADF9; AYLEAIHYF, AYF9; TLPEEFHDF, TDF9; TLPEEFHEF, TEF9; VCVAKLHDI, VCI9; VYVAKLHDI, VYI9; RYAALRELI, RI9; HYAALRELI, HI9) with >80% purity, and an HLA-A*24:02-bound conditional peptide sensitive to UV-light (VYG(X)VRACL, X as 3-amino-3-(2-nitro)-phenylpropionic acid)Citation58 with >95% purity, were purchased from Cosmo Bio (Tokyo, Japan). For HLA-A24 tetramer generation, biotinylated soluble HLA-A*24:02 monomers complexed with the conditional peptide sensitive to UV light and β2 m were synthesized by MBL (Nagoya, Japan). UV-mediated HLA-ligand exchange has been described previously.Citation59 Briefly, 28.8 μg/mL of the UV-sensitive monomers were mixed with 50 μM of peptides of interests in 96-well plates. For HLA-ligand exchange, the conditional ligand was cleaved upon UV illumination at 365 nm for 60 min on ice using a UV-Crosslinker CL-1000 L (Analytik Jena, US). The supernatant was transferred to a fresh tube after centrifugation at 3,300 × g for 5 min, mixed with 30 µg/mL streptavidin-R-PE (Invitrogen), and used for the assay after incubation for 60 min. The exchange efficiency of each ligand was measured using a sandwich ELISA. UV-exchanged monomers diluted 1:100 in PBS were transferred into streptavidin microplates (Thermo) and immobilized for 1 h at 37°C. The plates were extensively washed with 0.01% Tween-PBS and incubated with 100 µL of HRP-conjugated anti-human β2m antibody (2M2, BioLegend) for 1 h at 37°C. The absorbance at 405 nm was measured after the addition of 100 µL of ABTS peroxidase substrate (KPL), followed by incubation for 15 min at 25°C.
CD8+ T-cell induction from PBMCs using autologous DCs
A naive CD8+ T-cell subset of HD-PBMCs was stimulated by autologous DCs pulsed with peptides, as previously described.Citation36 Briefly, the adherent cell fraction of monocytes was isolated from 5.0 × 107 PBMCs, and differentiated into DCs by culturing in complete AIM-V medium supplemented with 1% human serum, 10 ng/mL IL-4 (Cell Genix), and 800 IU/mL GM-CSF (Gentaur) for 2 days, followed by additional culture in complete AIM-V medium supplemented with an increased concentration of 1,600 IU/mL GM-CSF for 24 h. DCs were then pulsed with 2.5 µM of antigen peptides of interest and cultured in the presence of 10 ng/mL LPS for maturation. The phenotypes of immature and mature DCs were confirmed using flow cytometry (Figure S5A). At the same time, naive CD8+ T cells were isolated from 1.0–2.0 × 108 PBMCs using a CD8 untouched isolation kit (Miltenyi) and CD45RO and CD57 beads (Miltenyi), according to the manufacturer’s instructions. The purity of the naive CD8+ T-cell subset was confirmed, which accounted for >95% of the recovered cells (Figure S5B). Finally, naive CD8+ T cells were mixed with irradiated mature DCs pulsed with antigen peptides in a 4:1 ratio (Day 0) and cultured in AIM-V medium supplemented with 5% human serum and 30 ng/mL IL-21 (Cell Genix) for 72 h. The mixed cells were further cultured in AIM-V medium supplemented with 5% human serum, 5 ng/mL IL-15 (Miltenyi), and 5 ng/mL IL-7 (Cell Genix) for 11 days. Cells on days −1 and 11 were analyzed by flow cytometry using UV-exchanged tetramers.
Establishment of CD8+ T-cell clones
CD8+ T-cell clones were established as described elsewhere.Citation60 Neoantigen-HLA-A24 tetramer-positive CD8+ cells were sorted using a FACS Aria II (BD Biosciences) and plated into 96-well plates as single cells. The cells were co-cultured with 1.0 × 105 irradiated allogeneic feeder cells in 200 µL of complete AIM-V medium supplemented with 10% human serum, 1 mM sodium pyruvate, 1% GlutaMAX, 55 µM 2-mercaptoethanol, 10 mM HEPES, 1% penicillin-streptomycin, 5 μg/mL phytohemagglutinin (Wako Chemicals), and 100 IU/mL IL-2.
Peptide-HLA class I stability assay
TAP-deficient T2-A24 cells were pre-cultured overnight at 25°C to increase the amount of empty HLA class I molecules on the cell surface. The next day, T2-A24 was incubated with peptides in a range of concentrations (0.3 to 3 µM) for 1 h at 25°C, followed by incubation for 3 h at 37°C. Stable peptide-HLA-A24 complexes remaining on the surface were then stained with an antibody specific to HLA-A24 (C7709A2) and goat anti-mouse IgG-FITC (KPL). Stability was calculated as the difference in mean fluorescence intensity (MFI) values between samples with and without a peptide pulse (ΔMFI) using a FACS Canto II (BD).
IFNγ ELISpot assay
Human IFNγ ELISpot plates (BD Biosciences) were prepared according to the manufacturer’s instructions. Antigen-presenting cells (T2 or T2-A24) were pre-incubated with 20 µM synthetic peptides for 2 h at 25°C. Fifty-thousand T cells were cultured with an equal number of antigen-presenting cells or cancer cells, on the plate for 24 h at 37°C. After the addition of 2 µg/mL biotinylated anti-human IFNγ antibody and streptavidin-HRP, reactive spots producing IFNγ were visualized using the AEC Substrate Set (BD Biosciences).
Intracellular IFNγ staining using flow cytometry
Fixation/Permeabilization Solution Kit with BD GolgiPlug (BD) was used according to the manufacturer’s instructions. Briefly, T2-A24 cells were pulsed with 20 µM of the indicated synthetic peptides for 1.5 h at 25°C and served as antigen-presenting cells. The indicated T-cell clones were incubated with T2-A24 cells at a 2:1 ratio for 4 h at 37 °C in the presence of anti-CD107a-PE (BioLegend), GolgiPlug, and human FcR blocking reagent (Clear Back, MBL). The cells were stained with anti-CD8 PC5, followed by permeabilization using a permeabilization solution (BD). The cells were subsequently stained with anti-IFNγ FITC and analyzed using FACS Canto II (BD).
Modeling of peptide-HLA structures and evaluation of physicochemical properties
The template crystal structure of a 9-mer peptide complexed with HLA-A*24:02 was obtained from the Protein Data Bank (PDB ID: 2BCK, https://www.rcsb.org). The peptide in the crystal structure was mutated to AKF9 or its 9-mer variants using PyMOL (Schrödinger). In the mutagenesis process, the most frequently occurring rotamers selected by default settings were chosen. Peptide-HLA docking was further optimized using the Rosetta FlexPepDock web server.Citation61 The obtained 3D models of neoantigen-HLA-A*24:02 complexes were visualized using PyMOL. To calculate the ASA of a given peptide, the PEP-FOLD3.5 web server was employed with default parameters and 100 simulation runs per peptide.Citation42 Physicochemical properties (molecular weight, net charge with the EMBOSS pKscale, and hydrophobicity index with the Kyte-Doolittle scale) of peptides were computed using the R package ‘Peptides’.Citation62 NetMHC %Rank scores were calculated using the online server NetMHCpan-4.1a (http://www.cbs.dtu.dk/services/NetMHCpan/index_4.1a.php).Citation27The PAM1 values were determined as previously reported.Citation63
Author contributions
TS performed and interpreted the experiments and wrote the manuscript. ST performed the HLA ligandome analysis and analyzed the data. ST and MN developed a proteogenomic pipeline for neoantigen detection. YH interpreted the experiments. TK conceived and designed the project, interpreted the experiments, and wrote the manuscript. TT supervised the project. All authors reviewed and approved the contents of the manuscript.
Competing interests
TK received grant support from Astellas and Eli Lilly. TT received grant support from Ono Pharmaceutical and Sumitomo Dainippon Pharma.
Supplemental Material
Download ()Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–11. doi:10.1126/science.aaa4971.
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371(23):2189–2199. doi:10.1056/NEJMoa1406498.
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348(6230):124–128. doi:10.1126/science.aaa1348.
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Geukes Foppen MH, Goldinger SM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–211. doi:10.1126/science.aad0095.
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 Blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372(26):2509–2520. doi:10.1056/NEJMoa1500596.
- Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413. doi:10.1126/science.aan6733.
- Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 2013;19(6):747–752. doi:10.1038/nm.3161.
- Lu YC, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, Davis L, Dudley ME, Yang JC, Samuels Y, et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res 2014;20(13):3401–3410. doi:10.1158/1078-0432.CCR-14-0433.
- Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, Gartner JJ, Zheng Z, Li YF, Ray S, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015;350(6266):1387–1390. doi:10.1126/science.aad1253.
- Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. T-Cell transfer therapy targeting mutant KRAS in Cancer. N Engl J Med 2016;375(23):2255–2262. doi:10.1056/NEJMoa1609279.
- Zacharakis N, Chinnasamy H, Black M, Xu H, Lu Y-C, Zheng Z, Pasetto A, Langhan M, Shelton T, Prickett T, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. 2018;6. doi:10.1038/s41591-018-0040-8
- Parkhurst MR, Robbins PF, Tran E, Prickett TD, Gartner JJ, Jia L, Ivey G, Li YF, El-Gamil M, Lalani A, et al. Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discov 2019;9(8):1022–1035. doi:10.1158/2159-8290.CD-18-1494.
- Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014;515(7528):572–576. doi:10.1038/nature14001.
- Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515(7528):577–581. doi:10.1038/nature13988.
- Bassani-Sternberg M, Braunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, Straub M, Weber J, Slotta-Huspenina J, Specht K, et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun 2016;7(1):13404. doi:10.1038/ncomms13404.
- Kalaora S, Barnea E, Merhavi-Shoham E, Qutob N, Teer JK, Shimony N, Schachter J, Rosenberg SA, Besser MJ, Admon A, et al. Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. Oncotarget 2016;7(5):5110–5117. doi:10.18632/oncotarget.6960.
- Kochin V, Kanaseki T, Tokita S, Miyamoto S, Shionoya Y, Kikuchi Y, Morooka D, Hirohashi Y, Tsukahara T, Watanabe K, et al. HLA-A24 ligandome analysis of colon and lung cancer cells identifies a novel cancer-testis antigen and a neoantigen that elicits specific and strong CTL responses. Oncoimmunology 2017;6(4):e1293214. doi:10.1080/2162402X.2017.1293214.
- Kalaora S, Wolf Y, Feferman T, Barnea E, Greenstein E, Reshef D, Tirosh I, Reuben A, Patkar S, Levy R, et al. Combined analysis of antigen presentation and T-cell Recognition reveals restricted immune responses in melanoma. Cancer Discov 2018;8(11):1366–1375. doi:10.1158/2159-8290.CD-17-1418.
- Ebrahimi-Nik H, Michaux J, Corwin WL, Keller GL, Shcheglova T, Pak H, et al. Mass spectrometry driven exploration reveals nuances of neoepitope-driven tumor rejection. JCI Insight. 2019;4(14):129152.doi:10.1172/jci.insight.129152.
- Loffler MW, Mohr C, Bichmann L, Freudenmann LK, Walzer M, Schroeder CM, Trautwein N, Hilke FJ, Zinser RS, Muhlenbruch L, et al. Multi-omics discovery of exome-derived neoantigens in hepatocellular carcinoma. Genome Med 2019;11(1):28. doi:10.1186/s13073-019-0636-8.
- Newey A, Griffiths B, Michaux J, Pak HS, Stevenson BJ, Woolston A, Semiannikova M, Spain G, Barber LJ, Matthews N, et al. Immunopeptidomics of colorectal cancer organoids reveals a sparse HLA class I neoantigen landscape and no increase in neoantigens with interferon or MEK-inhibitor treatment. Journal for Immunotherapy of Cancer 2019;7(1):309. doi:10.1186/s40425-019-0769-8.
- Freudenmann LK, Marcu A, Stevanovic S. Mapping the tumour human leukocyte antigen (HLA) ligandome by mass spectrometry. Immunology. 2018;154(3):331–345. doi:10.1111/imm.12936.
- Kanaseki T, Tokita S, Torigoe T. Proteogenomic discovery of cancer antigens: neoantigens and beyond. Pathol Int. 2019;9. doi:10.1111/pin.12841.
- Haen SP, Loffler MW, Rammensee HG, Brossart P. Towards new horizons: characterization, classification and implications of the tumour antigenic repertoire. Nat Rev Clin Oncol. 2020;10. doi:10.1038/s41571-020-0387-x.
- Abelin JG, Keskin DB, Sarkizova S, Hartigan CR, Zhang W, Sidney J, Stevens J, Lane W, Zhang GL, Eisenhaure TM, et al. Mass spectrometry profiling of HLA-Associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity 2017;46(2):315–326. doi:10.1016/j.immuni.2017.02.007.
- Hongo A, Kanaseki T, Tokita S, Kochin V, Miyamoto S, Hashino Y, Codd A, Kawai N, Nakatsugawa M, Hirohashi Y, et al. Upstream position of proline defines Peptide–HLA Class I Repertoire Formation and CD8+T Cell Responses. J Immunol. 2019;10. doi:10.4049/jimmunol.1900029
- Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48:W449–W54. doi:10.1093/nar/gkaa379.
- Fritsch EF, Rajasagi M, Ott PA, Brusic V, Hacohen N, Wu CJ. HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol Res. 2014;2(6):522–529. doi:10.1158/2326-6066.CIR-13-0227.
- Duan F, Duitama J, Al Seesi S, Ayres CM, Corcelli SA, Pawashe AP, Blanchard T, McMahon D, Sidney J, Sette A, et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med 2014;211(11):2231–2248. doi:10.1084/jem.20141308.
- Capietto AH, Jhunjhunwala S, Pollock SB, Lupardus P, Wong J, Hansch L, Cevallos J, Chestnut Y, Fernandez A, Lounsbury N, et al. Mutation position is an important determinant for predicting cancer neoantigens. J Exp Med. 2020;(4):217. doi:10.1084/jem.20190179.
- Gattoni-Celli S, Kirsch K, Timpane R, Isselbacher KJ. Beta 2-microglobulin gene is mutated in a human colon cancer cell line (HCT) deficient in the expression of HLA class I antigens on the cell surface. Cancer Res. 1992;52:1201–1204.
- Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61. doi:10.1016/j.cell.2014.12.033.
- Inoda S, Hirohashi Y, Torigoe T, Morita R, Takahashi A, Asanuma H, Nakatsugawa M, Nishizawa S, Tamura Y, Tsuruma T, et al. Cytotoxic T lymphocytes efficiently recognize human colon cancer stem-like cells. Am J Pathol 2011;178(4):1805–1813. doi:10.1016/j.ajpath.2011.01.004.
- Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteomics. 2015;14(3):658–673. doi:10.1074/mcp.M114.042812.
- Stronen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, Donia M, Boschen ML, Lund-Johansen F, Olweus J, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. 2016;6291. doi:10.1126/science.aaf2288
- Wolfl M, Greenberg PD. Antigen-specific activation and cytokine-facilitated expansion of naive, human CD8+ T cells. Nat Protoc. 2014;9(4):950–966. doi:10.1038/nprot.2014.064.
- Harndahl M, Rasmussen M, Roder G, Dalgaard Pedersen I, Sorensen M, Nielsen M, Buus S. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur J Immunol 2012;42(6):1405–1416. doi:10.1002/eji.201141774.
- Luksza M, Riaz N, Makarov V, Balachandran VP, Hellmann MD, Solovyov A, Rizvi NA, Merghoub T, Levine AJ, Chan TA, et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017;551(7681):517–520. doi:10.1038/nature24473.
- Rech AJ, Balli D, Mantero A, Ishwaran H, Nathanson KL, Stanger BZ, Vonderheide RH. Tumor immunity and survival as a function of alternative neopeptides in human cancer. Cancer Immunol Res. 2018;3. 10.1158/2326-6066.CIR-17-0559
- Ghorani E, Rosenthal R, McGranahan N, Reading JL, Lynch M, Peggs KS, Swanton C, Quezada SA. Differential binding affinity of mutated peptides for MHC class I is a predictor of survival in advanced lung cancer and melanoma. Annals of Oncology: Official Journal of the European Society for Medical Oncology. 2018;29:271–279. doi:10.1093/annonc/mdx687.
- Riley TP, Keller GLJ, Smith AR, Davancaze LM, Arbuiso AG, Devlin JR, Baker BM. Structure based prediction of neoantigen immunogenicity. Front Immunol. 2019; pp.10. doi:10.3389/fimmu.2019.02047
- Lamiable A, Thevenet P, Rey J, Vavrusa M, Derreumaux P, Tuffery P. PEP-FOLD3: fasterde novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016;44(W1):W449–54. doi:10.1093/nar/gkw329.
- Chowell D, Krishna S, Becker PD, Cocita C, Shu J, Tan X, Greenberg PD, Klavinskis LS, Blattman JN, Anderson KS. TCR contact residue hydrophobicity is a hallmark of immunogenic CD8 T cell epitopes. Proc Natl Acad Sci U S A 2015;112(14):E1754–62. doi:10.1073/pnas.1500973112.
- Teku GN, Vihinen M. Pan-cancer analysis of neoepitopes. Sci Rep. 2018;8(1):12735. doi:10.1038/s41598-018-30724-y.
- Sewell AK. Why must T cells be cross-reactive? Nat Rev Immunol. 2012;12(9):669–677. doi:10.1038/nri3279.
- Zhang SQ, Ma KY, Schonnesen AA, Zhang M, He C, Sun E, Williams CM, Jia W, Jiang N. High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat Biotechnol. 2018;12. 10.1038/nbt.4282
- Bentzen AK, Such L, Jensen KK, Marquard AM, Jessen LE, Miller NJ, Church CD, Lyngaa R, Koelle DM, Becker JC, et al. T cell receptor fingerprinting enables in-depth characterization of the interactions governing recognition of peptide-MHC complexes. Nat Biotechnol. 2018. doi:10.1038/nbt.4303.
- Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, Wong YNS, Rowan A, Kanu N, Bakir MA, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol 2017;18(8):1009–1021. doi:10.1016/S1470-2045(17)30516-8.
- Mandal R, Samstein RM, Lee KW, Havel JJ, Wang H, Krishna C, Sabio EY, Makarov V, Kuo F, Blecua P, et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 2019;364(6439):485–491. doi:10.1126/science.aau0447.
- Leng Q, Tarbe M, Long Q, Wang F. Pre-existing heterologous T-cell immunity and neoantigen immunogenicity. Clin Transl Immunol. 2020;9:e1111. doi:10.1002/cti2.1111.
- Fluckiger A, Daillere R, Sassi M, Sixt BS, Liu P, Loos F, Richard C, Rabu C, Alou MT, Goubet AG, et al. Cross-reactivity between tumor MHC class I-restricted antigens and an enterococcal bacteriophage. Science 2020;369(6506):936–942. doi:10.1126/science.aax0701.
- Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, Herbst B, Askan G, Bhanot U, Senbabaoglu Y, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017;551(7681):512–516. doi:10.1038/nature24462.
- Wells DK, van Buuren MM, Dang KK, Hubbard-Lucey VM, Sheehan KCF, Campbell KM, Lamb A, Ward JP, Sidney J, Blazquez AB, et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell. 2020);3. 10.1016/j.cell.2020.09.015
- Scholtalbers J, Boegel S, Bukur T, Byl M, Goerges S, Sorn P, Loewer M, Sahin U, Castle JC. TCLP: an online cancer cell line catalogue integrating HLA type, predicted neo-epitopes, virus and gene expression. Genome Med 2015;7(1):118. doi:10.1186/s13073-015-0240-5.
- Nakatsugawa M, Hirohashi Y, Torigoe T, Inoda S, Kiriyama K, Tamura Y, Sato E, Takahashi H, Sato N. Comparison of speedy PCR-ssp method and serological typing of HLA-A24 for Japanese cancer patients. J Immunoassay Immunochem 2011;32(2):93–102. doi:10.1080/15321819.2010.543219.
- Kowalewski DJ, Stevanovic S. Biochemical large-scale identification of MHC class I ligands. Methods Mol Biol. 2013;960:145–157.
- Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res 2017;45(D1):D777–D83. doi:10.1093/nar/gkw1121.
- Chang CX, Tan AT, Or MY, Toh KY, Lim PY, Chia AS, Froesig TM, Nadua KD, Oh HLJ, Leong HN, et al. Conditional ligands for Asian HLA variants facilitate the definition of CD8+ T-cell responses in acute and chronic viral diseases. Eur J Immunol 2013;43(4):1109–1120. doi:10.1002/eji.201243088.
- Rodenko B, Toebes M, Hadrup SR, van Esch WJ, Molenaar AM, Schumacher TN, Ovaa H. Generation of peptide–MHC class I complexes through UV-mediated ligand exchange. Nat Protoc. 2006;1(3):1120–1132. doi:10.1038/nprot.2006.121.
- Miyamoto S, Kochin V, Kanaseki T, Hongo A, Tokita S, Kikuchi Y, Takaya A, Hirohashi Y, Tsukahara T, Terui T, et al. The antigen ASB4 on cancer stem cells serves as a target for CTL immunotherapy of colorectal cancer. Cancer Immunol Res. 2018;6(3):358–369. doi:10.1158/2326-6066.CIR-17-0518.
- London N, Raveh B, Cohen E, Fathi G, Rosetta FlexPepDock S-FO. web server–high resolution modeling of peptide-protein interactions. Nucleic Acids Res. 2011;39(suppl_2):W249–53. doi:10.1093/nar/gkr431.
- Osorio D, Rondon-Villarreal P, Torres R. Peptides: A Package for Data Mining of Antimicrobial Peptides. The R Journal 2015; 7
- Dayhoff MO, Schwartz RM, Orcutt BC. Chapter 22: A model of evolutionary change in proteins. In Atlas of protein sequence and structure. 1978; Vol. 5. 345–352