
ABSTRACT
The immune system can recognize tumor cells to mount antigen-specific T cell response. Central to the establishment of T cell-mediated adaptive immunity are the inflammatory events that facilitate antigen presentation by stimulating the expression of MHC and costimulatory molecules and the secretion of pro-inflammatory cytokines. Such inflammatory events can be triggered upon cytotoxic treatments that induce immunogenic cancer cell death modalities. However, cancers have acquired a plethora of mechanisms to subvert, or to hide from, host-encoded immunosurveillance. Here, we discuss how tumor intrinsic oncogenic factors subvert desirable intratumoral inflammation by suppressing immunogenic cell death.
Introduction
The tumor microenvironment comprises malignant cells, immune cells (innate and adaptive), structural cells (e.g. fibroblasts), vascular cells and others. These cellular constituents function as ecosystems in thus far that they exhibit cooperation (e.g. reciprocal provision of growth factors), competition (e.g. for oxygen, nutrients), predation (e.g. antitumor immunity), and co-evolution in response to selective pressure. The immune system exerts complex tasks to eliminate growing tumors. Innate immune cells such as natural killer (NK) cells are the first to encounter and fight off cancer and, if tumors are not eliminated, the adaptive immune system takes over to mount tumor-specific response by engaging cytotoxic T lymphocytes (CTLs) which are endowed with receptors that recognize tumor antigens presented in the context of major histocompatibility complex (MHC) (). Cancer immunoediting is the process whereby the immune system can both constrain and promote tumor growth. The concept of cancer immunoediting relies on sequential stages, namely elimination, equilibrium and escape, that depict the interaction of growing tumors with the immune systemCitation1. Over-expressed tumor-associated self-antigens, non-self-mutated neoantigens and oncogenic virus-associated epitopes can be cross-presented to naïve T cells and activate tumor-specific CTL responses to eliminate immunogenic tumorsCitation1. The immune pressure added to genomic instability provides tumors an opportunity to develop malignant escape variants that undergo progressive selection and outgrowth. Thus, clinically diagnosed macroscopic tumors are usually immune-edited and have defeated host intrinsic immunosurveillance mechanisms ().
Figure 1. Mechanisms of tumor antigen presentation
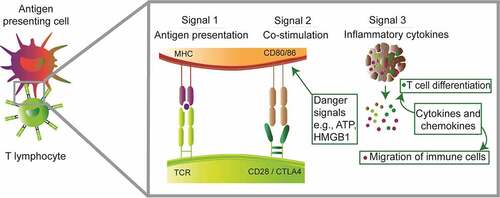
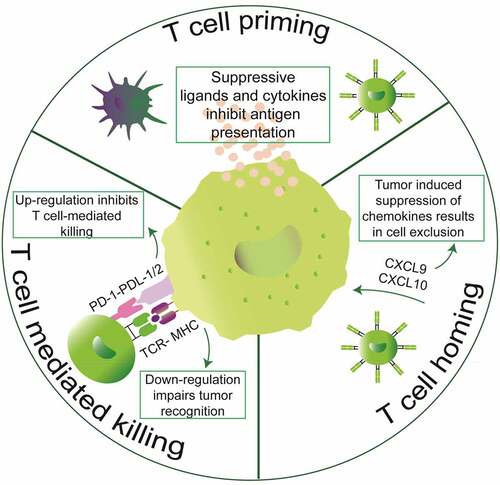
Figure 2. Mechanisms of tumor-mediated immune evasion. Tumors can inhibit intrinsically developed immunity at the three stages of anticancer immunity cycle. Tumors secrete suppressive cytokines and ligands to inhibit T cell priming. Moreover, tumors can also conspire the secretion of chemokines that are essential for entry of cytotoxic T lymphocytes into the tumor stroma. Lastly, tumors can also inhibit T cell-mediated killing either by downregulating components of the antigen processing and presentation machinery or by expressing T cell inhibitory ligands such as PD-L1/2
Several immunotherapeutic approaches can reactivate preexisting immune infiltrates to provide long-term survival of cancer patients. Moreover, in tumors that lack immune infiltrates induction of inflammatory cell death can kickstart the anticancer immunity cycle and reinstate immunosurveillance ().Citation2 In fact, certain types of cytotoxic chemotherapiesCitation3 or targeted therapies such as radiation,Citation4 oncolytic viruses,Citation5 photodynamic therapy,Citation6 extracorporeal photochemotherapyCitation7,Citation8 can activate tumor cell stress and immunogenic cell death (ICD) that positively contributes to immune-mediated recognition of tumorsCitation3 (). Depending on the cytotoxic stimuli, ICD consists of a cascade of events that starts with a premortem stress and leads to a cellular demise that concurrently allows the release of immunomodulatory molecules and tumor cell contents. In these scenarios, danger molecules and cytokines released during ICD are critical for successful tumor-antigen presentation and development of adaptive immunity. Thus, approaches that combine ICD inducers with immune checkpoint inhibitorsCitation2 that block inhibitory T cell receptorsCitation9,Citation10 have gained clinical popularity.Citation11,Citation12 However, emerging studies show direct evidence that tumor-intrinsic oncogenic factors can influence the immunogenicity of tumor cell deathCitation13,Citation14 (). Here we review recent advances as to how tumor-specific oncogenic factors (oncogenes and tumor suppressors) influence the anticancer immunity cycle by dictating the activation of inflammatory cell death as well as the secretion of immunomodulatory molecules during ICD. We conclude by illustrating how knowledge of tumor genetics can be integrated into patient selection for maximizing immunotherapeutic outcome after ICD-inducing treatments.
Figure 3. Mechanisms of ICD. Several types of lethal stimuli (a) activate tumor cell stress and cell death that leads to the surface expression of the “eat me” signal calreticulin and extracellular release of ATP, HMGB1 and interferons (b). In a concerted effort, the danger molecules released during ICD promote antigen presentation and immune cell trafficking (c)
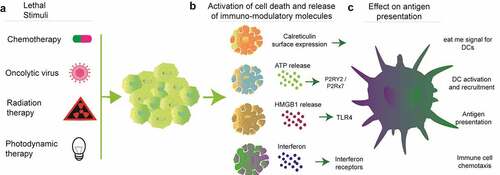
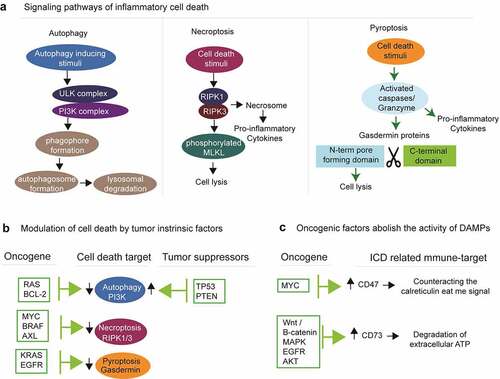
Figure 4. Tumor intrinsic oncogenic factors dictate the activation and execution of ICD. (a) Diverse genotoxic and metabolic stimuli initiate signaling pathways to activate autophagy, necroptosis and pyroptosis. (b) All these three types of inflammatory cell death can be modulated by tumor intrinsic factors. (c) Several types of oncogenes evade ICD by indirectly activating immune-suppressive ligands
Mechanisms of antitumor immunity after ICD
Mechanisms of ICD and downstream events leading to tumor-specific CTL response have been mostly investigated in the context of cancer chemotherapy. In this setting, stressed and dying malignant cells display or release Danger Associated Molecular Patterns (DAMPs) that attract and activate proficient antigen-presenting cells, mainly immature dendritic cells (DCs). Among these alarm signals are (1) the surface exposure of the endoplasmic reticulum chaperone calreticulin (CALR) and the release into the tumor microenvironment (TME) of (2) the cytoplasmic protein annexin A1 (ANXA1), (3) the nucleotide ATP, and (4) the nonhistone chromatin-binding protein high-molecular group B1 (HMGB1) (). These DAMPs are sensed by DCs through the low-density lipoprotein receptor-related protein 1 (LRP1, also known as CD91), formyl peptide receptor-1 (FPR1), purinergic receptors P2RX7 and P2RY2, and TLR4, respectively. Additionally, treatment by ICD chemo-inducers mimics viral infection and stimulates the secretion by malignant entities of type I interferons (IFN) and C-X-C motif chemokine ligand 10 (CXCL10), thus attracting T lymphocytes. Once at proximity of DAMP-emitting dying cancer cells, DCs will experience activation and maturation which implies up-regulation of the lymphoid tissue-residing C-C motif chemokine receptor 7 (CCR7), of MHC molecules and co-stimulatory factors (e.g. CD80, CD86), as well as the production of inflammatory cytokines (e.g. interleukin-12 [IL12], IL6, tumor necrosis factor-alpha [TNF-α]).
DCs are able to engulf tumor antigens notably via phagocytosis of malignant cell corpses, or via macropinocytosis of free antigens that may have spread upon cell death.Citation15 Then, DCs process captured antigens and proceed to cross-presentation of associated epitopes onto MHC (). Of note, DC populations that appeared particularly enriched in the tumor bed and seemed predictive of an improved cancer outcome consist of Ly6ChiCD11b+ monocyte-derived DCs and of Clec9A+ type 1 conventional DCs, particularly the CD103+ subset.Citation16 Recently, a platelet factor P-selectin was shown to initiate cross-presentation.Citation17 Mature DCs migrate to secondary lymphoid organs to prime cognate naïve CD4+ and CD8+ T lymphocytes.Citation18 This step not only requires the interaction of the MHC/epitope complex with the T cell receptor (TCR) but also a co-stimulatory signal resulting from the binding of DC surface-exposed CD80/CD86 to T cell surface-exposed CD28, as well as the secretion of some cytokines supporting T cell differentiation and proliferation such as IL2Citation19(). Activated type 1 helper CD4+ T (Th1) and cytotoxic CD8+ T (Tc1) lymphocytes also produce large amounts of IFNγ, which exhibits pleiotropic antitumor activity. First, IFNγ maintains Th1 T cell lineage commitment and stimulates differentiation of CD8+ T cells into CTLs (positive feedback loop), boosts antigenic exposure in antigen-presenting cells and target cells, and favors macrophage activation and polarization toward a tumoricidal phenotype (e.g. enhanced phagocytic potential, production of nitric oxide, tryptophan depletion)20. Second, IFNγ can exert direct anti-proliferative (e.g. regulation of p21 expression), anti-angiogenic (e.g. impaired survival of endothelial cells) and pro-apoptotic/necroptotic (e.g. up-regulation of caspases, enhanced secretion of Fas and Fas ligand) effects on transformed cells.Citation20 Additionally, activated T lymphocytes produce TNFα which cooperates with IFNγ to further stimulate Th1/Tc1 responses and to sensitize cancer cells to apoptosis (e.g. repression of B-cell lymphoma-extra large [Bcl-xL] expression, ischemia via endothelial cell apoptosis).Citation20,Citation21 Upon activation, effector T cells, notably CTLs, express C-X-C motif chemokine receptor 3 (CXCR3) that will allow their migration to the tumor bed in a, yet poorly understood, paracrine CXCL9/CXCL10/CXCL11-dependent mannerCitation22 (). These chemokines are mostly secreted by cancer cells, monocytes, endothelial cells and fibroblasts in response to IFN-γCitation22 Then malignant cells harboring MHC-I-coupled epitopes can be targeted by cognate CTLs which achieve their antitumor function mainly through the secretion of perforin, a pore-forming toxin triggering osmotic lysis.Citation23,Citation24 Collectively, this series of events has been termed “cancer-immunity cycle” by Dan Chen and Ira Mellman back in 2013Citation18 ().
Adaptive immune-resistance mechanisms encoded by tumors
Cancer cells adapt to overcome the aforementioned immunosurveillance mechanisms and resist to immune attack. These pro-tumoral immune evasion actions mostly prevent either the ignition of the cancer-immunity cycle at the level of antigen presentation by inhibiting DC function, or terminally suppress the effector function of cytotoxic T lymphocytesCitation25 (). First, immunoediting tends to select poorly immunogenic transformed cells. Hiding from the adaptive immune cell radar can be achieved through mutations or epigenetic silencing of relevant tumor antigens or through a general reduction of antigen presentation, via the loss of class-I MHC, beta-2-microglobulin (B2M) or antigen peptide transporters 1 and 2 (TAP1/2).Citation26,Citation27 Alternatively, some malignant cells constitutively expose inhibitory immune checkpoints such as programmed death-ligand 1 (PD-L1)Citation28 or secrete immunosuppressive factors like indoleamine 2, 3-dioxygenase 1 (IDO1).Citation29 Both PD-L1 and IDO1 are normally stimulated by IFN-γ as a negative feedback loop.Citation29–31 PD-L1 interacts with the activation/exhaustion marker programmed cell death 1 (PD-1) on tumor-infiltrating T lymphocytes (TILs) and impair cytokine production as well as T cell proliferation and survival.Citation32 Released IDO1 depletes tryptophan (Trp) from the extracellular milieu thus promoting cell cycle arrest and death of effector TILs.Citation29 Furthermore, Trp catabolism by IDO1 enzymatic activity generates kynurenine (Kyn). The latter can bind the aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor, at the surface of DCs and regulatory CD4+ T lymphocytes (Tregs). In DCs, Kyn-activated AhR triggers the release of immunosuppressive IL10 while inhibiting the production of immunostimulatory IFNβ. Meanwhile, nuclear translocation of activated AhR in immunosuppressive Tregs stimulates their proliferation within the TME.Citation29 In addition to IFN-induced adaptive immune resistance, cancer cells also acquire resistance to cancer immunosurveillance and immunotherapy via inflammatory cytokine- and stress-related mechanisms.Citation33–36 For instance, in a preclinical murine model of melanoma, the release of TNFα by TILs initiated de-differentiation of melanoma cells and ultimately translated into a loss of several melanosomal antigens. As a consequence, a therapy relying on the adoptive transfer of T cells specific for the melanoma-associated antigen gp100 only showed transient responses.Citation33,Citation34 Oncogenic pathways that support the abovementioned mechanisms of adaptive immune resistance remain poorly documented and will be introduced in the following paragraph.Citation37
Oncogenic driver mutations influence cancer immune contexture and immunosurveillance
Genomic landscapes of common human cancers revealed around 140 genes that can drive tumorigenesis when altered by intragenic mutations.Citation38 Common to most solid tumors is that the major driver oncogenic mutations confer selective growth and proliferative advantage by altering up to 12 cancer cell-intrinsic cellular signaling pathways.Citation38 These deregulated pathways confer distinct and complementary capabilities that confer on tumor cells a selective growth advantage by increasing cell division and preventing cell death.Citation39 In parallel, cancer cells must acquire the ability to prevent immune cell recognition and elimination.Citation40 The composition and function of immune cells in tumors vary greatly between and within cancer histotypes.Citation41,Citation42 This considerable variation in phenotypic and functional characteristics of intratumoral leukocyte composition impact cancer outcome.Citation43 Accumulating clinical observations and mouse studies reveal that gain-of-function mutations in oncogenes (e.g. KRAS, MYC) and loss-of-function alterations in tumor suppressor genes (e.g. PTEN, TP53) are correlated with changes in immune composition and response to immunotherapy.Citation44 Overall, mouse and human studies showed that inactivation of tumor suppressors as well as activation of oncogenes directly contribute to pro-tumoral immune contexture and failed immunosurveillance.Citation44 The mechanisms by which oncogenic driver mutations orchestrate highly immune-suppressive tumor microenvironments are under intense investigation. Pro-oncogenic signals can affect the cancer-immunity cycle at the stages of (1) tumor antigen capture and presentation, (2) T cell activation and recruitment to malignant lesions, and of the (3) tumoricidal activity of T cells within the tumor (). This review will document how tumor intrinsic oncogenic events influence ICD and thereby influence tumor antigen presentation.
Do tumor-intrinsic oncogenic events affect ICD?
Over-activated oncogenic signaling at early stages of cellular transformation creates metabolic stress that compel malignant cells to activate programmed cell death.Citation45 Thus, only malignant cells that acquire additional molecular defects to confer interruption of cell death signaling cascades are able to continue proliferation.Citation46,Citation47 In addition to cell-autonomous effects, cell death effectors can exert non-cell-autonomous effects by regulating the release of danger molecules and cytokines into the tumor microenvironment.Citation48–50 Thus, tumors manipulate programmed cell death to evade immunosurveillance. Malignant cells suppress programmed cell death signaling through loss-of-function mutations in proteins that sense and transduce lethal signals or execute cell death (as in many tumor suppressor proteins), and also from gain-of-function alterations in oncogenes that normally deliver pro-survival signals.Citation51,Citation52 Depending on the cytotoxic stimuli, ICD consists of a cascade of events which starts with a premortem stress and leads to a cellular demise that concurrently allows the release of immunomodulatory molecules and tumor cell content. Thus, successful execution of ICD involves three distinct and interlinked biological processes that include (1) activation of premortem stress and execution of inflammatory cell death, (2) expression and/or secretion of immunomodulatory molecules, and (3) exogenous effects of these immunomodulatory molecules which consist in the recruitment and activation of immune cells (). In the next section of the review, we provide new insights and mechanisms as to how tumor-specific oncogenic events interfere at the first two steps of ICD activation.
1. Effect of tumor-intrinsic oncogenic events on the initiation and execution of inflammatory cell death modalities
It is now widely accepted that oncogenic events, depending on cancer type, select for tumors that initially resist cell death. Due to inactivation of the molecular pathways that sense cellular stress to activate cell death, malignant cells are relatively resistant to death upon chemotherapy and radiotherapy.Citation53 Although the mechanisms are mostly unknown, tumors dampen signaling pathways required for sensing intracellular and extracellular stress.Citation54,Citation55 Several sensors of damage-associated molecular patterns (DAMPs), including Toll-like receptors (TLRs),Citation55 the cytosolic RNA-sensors retinoic acid-inducible gene I (RIG-I)Citation56 and melanoma differentiation A protein 5 (MDA-5), as well as stimulator of interferon genes (STING),Citation54 appear down-regulated in multiple cancers compared to healthy tissues. Collectively, these deregulated pathways enable tumor cells to counter immunosurveillance, partly by preventing them to sense stress and to activate immune sentinels. Recent studies showed that oncogenic factors interfere in certain tumor types with the activation and execution of inflammatory cell death by regulating the expression of cell death executioner caspasesCitation57,Citation58 and kinasesCitation14 (). Moreover, inhibition of major oncogenes, such as KRAS, causes tumors to die in an immunogenic fashion thereby reinstating immunosurveillance.Citation13 The following sections of the review will describe how tumor-intrinsic oncogenic factors interfere with ICD by manipulating cell death and survival programs including autophagy, necroptosis and pyroptosis, or again ICD-related hallmarks.
Autophagy
Autophagy is an evolutionarily conserved lysosomal degradation process that is critical for nutrient recycling and metabolic adaptation during stress (). Thus, autophagy intervenes in a plethora of biological phenomena such as mitochondrial function, stress response, cellular homeostasis, metabolism, cell death and immune surveillance.Citation59 Autophagy is linked to many types of pathologies including cancer. Autophagy plays a complex role in tumor development and progression by influencing different aspects of the tumor–host interaction. Autophagy constitutes a cell-intrinsic barrier against malignant transformationCitation60 by activating oncogene-induced cell death,Citation45 cellular senescence,Citation61,Citation62 removal of reactive oxygen species which can damage mitochondria,Citation63 maintenance of genomic stabilityCitation64 and degradation of oncogenic proteins.Citation65–69 Consistent with this notion, deletion of autophagy regulators in tumor-prone mouse results in increased incidence of lung and liver tumors as well as lymphomas.Citation70–73 However, in established tumors, autophagy is often used as an adaptive mechanism for tumors to thrive in nutrient-depleted and hypoxic tumor microenvironments.Citation74–77 Although tumor cells vary in their autophagy dependency, inhibiting autophagy in established tumors generally results in dramatic tumor reduction and prolonged survival in murine models.Citation78,Citation79 In accordance with these observations, advanced human tumors often display enhanced autophagic flux.Citation80
The non-cell-autonomous antitumor effects of autophagy are linked to its immunomodulatory role in the immune TME.Citation12,Citation81–83 Autophagy induction in anthracycline-treated dying tumors facilitates the release of immune-stimulatory ATP to the extracellular microenvironment, thereby potentiating tumor-antigen presentation and immune-mediated recognition of tumors.Citation81–84 In this line, deletion of ATG5 in KRAS mutant tumors results in accelerated oncogenesis by creating highly immunosuppressive microenvironment associated with Treg-mediated inhibition of cancer immunosurvillence.Citation78 Moreover, autophagy has been shown to promote cross-priming of tumor-specific CD8+ T cells.Citation85 In immune cells, autophagy plays a role in the formation of memory CD8+ T cells.Citation86 Overall, these findings suggest that both tumor-intrinsic and systemic host defects in autophagy may prevent the immune system to detect and eliminate pre-malignant and malignant cells. In contrast, tumor cell autophagy has also been shown to inhibit NK cell cytotoxicity in some tumor types.Citation87
Due to its tumor cell-autonomous and immune-mediated microenvironmental effects, autophagy is subject to modulation by oncogenic and tumor suppressor proteinsCitation88,Citation89 (). Many oncogenes, such as BCL-2 and RAS directly inactivate components of the autophagy machinery. Several members of the BCL-2 protein family inhibit autophagy by sequestering BECN1.Citation90–92 Hyperactivated RAS engages the PI3K/PDPK1/AKT1 signaling cascade to potently supress autophagic responses.Citation93 Tumor suppressors such as TP53 engage the cell death machinery in tumors that harbor irreparable molecular lesions such as DNA damage. TP53 can promote autophagy by transactivating multiple autophagy-related genes including AMPK and BH3 family of proteins (such as BAD, BNIP3). Moreover, the phosphatase and tensin homolog protein (PTEN) is a tumor suppressor with autophagy promoting functions by antagonizing PI3K signaling. The transcription factor forkhead box O1 (FOXO1) is a tumor suppressor required for stress-induced autophagy by directly interacting with ATG7. Taken together, tumor suppressors activate autophagy to limit malignant cell progression. Malignant cells that have acquired the hallmarks of inactivating tumor suppressors along with hyperactivated oncogenes will progress to down-regulate autophagy. Additional genetic defects may cooperate with oncogenes and tumor suppressors to regulate autophagy during tumor progression.
Necroptosis
Necroptosis is a programmed cell death triggered by various stimuli that include engagement of death receptors, IFNs, TLRs, intracellular RNA and DNA sensors, as well as genotoxic and oxidative stresses induced by anticancer drugsCitation94 (). The role of necroptosis in anticancer immunity remains unclear. Indeed, necroptosis can be pro-tumoralCitation95 and antitumoralCitation96–98 depending on the nature of the lethal stimulus and tumor model. On one side, George Miller and colleagues identified that pancreatic tumors have higher basal expression of the necrosome components that is further augmented after treatment with gemcitabine. Impeding necroptosis in vivo resulted in a more inflammatory tumor infiltrate associated with elevated CD8+ T lymphocytes and reduced myeloid-derived suppressor cells. These pro-tumorigenic effects of necroptosis seemed specific to tumors that were growing in the pancreatic microenvironment. Yet, recently in melanoma, an unbiased CRISPR screen identified RIPK1 as a top candidate inhibiting immunotherapy with immune checkpoint inhibitors.Citation99 On the other side, antitumor effects of necroptosis are based on overexpression of the executioner proteins RIPK3 and MLKL.Citation96–98 These observations were mainly made in vaccination studies that specifically activate necroptosis in fibroblasts following chemically induced dimerization of RIPK1/3 to activate cytokine secretion and cell lysis. Intratumoral injection of necroptotic fibroblasts provides pro-inflammatory cytokines that serve as adjuvants to activate antitumor immunity in a non-antigen-dependent fashion.Citation96–98 The paradoxical effects of necroptosis on immune activation may arise from differences in the lethal stimuli and immune responsiveness of the tumor models.
Components of the necroptosis machinery are deregulated in many cancer types (). Tumors display different types of mutations in the proteins that execute necroptosis. Some cancer types shut off necroptosis through (1) genetic and epigenetic down-regulation of RIPK3 and MLKL expression in acute myeloid leukemia, breast, colon and colorectal cancer types,Citation100–102 and (2) acquired mutations in functional domains of RIPK3 and MLKL that hinder necroptosis signaling or cell lysis during necroptosis.Citation103,Citation104 In this line, tumor-specific oncogenic events are shown to directly regulate the expression of RIPK1 and RIPK314. The actin crosslinking protein α-actinin-4 (ACTN4) is emerging as crucial regulator of carcinogenesis. ACTN4 serves as a scaffold to stabilize RIPK1 by allowing association of RIPK1 and cellular inhibitor of apoptosis protein 1 (cIAP1) to activate NF-κB.Citation105 A recent study on 941 human cancer cell lines came to the conclusion that 83% of the cells are resistant to necroptosis, irrespective of the tissue and cancer subtype. Bioinformatic analyses revealed that 20 oncogenic hits hinder necroptosis by down-regulating RIPK3 expression.Citation14 Chemical inhibition of the oncogenes AXL (using BMS-777607) and BRAF (TAK-632) up-regulated RIPK3 expression in tumor cells.Citation14 However, shutting down the necroptosis pathway is not a general mechanism exploited by all types of cancer cells to survive and progress. Indeed, the expression of necroptotic players was found to be elevated in glioblastoma,Citation106 lungCitation107 and ovarian cancers.Citation108 Future studies should explore the genetic and epigenetic interactions of oncogenes and tumor suppressors with the necroptosis machinery in a broad range of cancers.
Pyroptosis
Pyroptosis is an inflammatory form of programmed necrosis that serves as an immune effector mechanism against microbes and cancerCitation109 (). A diverse range of ligands and genotoxic stressors stimulate the inflammatory signaling cascade that culminates in the activation of caspases that subsequently cleave and activate gasdermin. Irrespective of the cell death stimuli and signaling cascade, gasdermin cleavage represents a terminal event during pyroptosis.Citation110 Proteins of the gasdermin family (which consists of gasdermins A, B, C, D, and E as well as of Pejvakin) are expressed in normal tissuesCitation111 in an autoinhibited state (with the exception of Pejvakin). Following caspase- and granzyme A/B-mediated cleavage, the active N-terminal domain of gasdermin binds to the plasma membrane to generate pores that disrupt their barrier function, resulting in cell swelling and eventual lysis.Citation111 Moreover, the pores serve as gates for the extracellular release of danger signals and cytokines.Citation111
The pathophysiological role of pyroptosis in cancer is expanding. Many types of anticancer chemotherapies including topotecan, etoposide, cisplatin, 5-fluoruracil and paclitaxel activate pyroptosis in cancer cell lines in a gasdermin-dependent fashion. Activation of pyroptosis in tumors can exert both tumor-promoting and antitumor immune effects.Citation112 Pro-tumor effects of pyroptosis are reported for pancreatic tumors and mainly linked to chronic activation of the inflammasome which attracts MDSCs.Citation113 The tumor-promoting role of inflammasomes is related to immune suppression consecutive to secretion of the cytokines IL-1βCitation114,Citation115 and IL-18.Citation116–118 On the contrary, antitumor effects of inflammasomes were witnessed in colorectal cancer. As inflammasomes are major drivers of pyroptosis, the lack of inflammasome mediators in colorectal cancer was associated with pronounced tumor growth.Citation119–122 In line with the antitumor effects of pyroptosis, loss of gasdermin expression is correlated with aggressive cancers and increased risk of metastasisCitation123 whereas expression of full-length gasdermin E in mouse cancers stimulate antitumor immunity.Citation124,Citation125 Two independent groups showed that ectopically expressed full-length gasdermin E is cleaved by granzyme A/B released by cytotoxic T lymphocytes and NK cells, resulting in the release of N-terminal gasdermin that forms pores in cancer cells.Citation124,Citation125
Several studies revealed that gasdermin proteins A, C, D and E are down-regulated in human breast,Citation125 gastricCitation126 and colorectal cancer cell lines, as well as in primary tumorsCitation126,Citation127 (). Accordingly, targeted therapies like erlotinib and trametinib, which inhibit oncogenic signaling by targeting EGFR and KRAS, respectively, resulted in increased pyroptosis.Citation128 These results suggest that the oncogenes EGFR and KRAS may suppress pyroptosis. Further research should explore the mechanisms by which tumor-specific oncogenic events downregulate pyroptosis.
2. Effect of tumor-intrinsic oncogenic events on the secretion of ICD-induced DAMPs
Sensing of lethal stimuli to activate cellular stress and programmed cell death is essential to enforce tumor cells to emit immunomodulatory molecules (). In the last two decades, the molecular mechanisms of ICD-induced immune activation have profoundly enriched our understanding on the immunomodulatory molecules released during cell deathCitation129,Citation130 and how that affects recruitment of immune cells to the tumor bed, innate and adaptive immune cell activation, and anticancer activity.Citation2,Citation82 Overall, these studies have shown that various forms of ICD inducers promote the exposure or release/secretion of immunomodulatory molecules such as CALR,Citation131 HMGB1, ATP,Citation132 type I IFNs,Citation41 CXCL10,Citation41 and CXCL1.Citation42 ICD inherently relies on the ability of cancer cells to display these signals (). Yet, tumors vary in their ability to secrete immuno-modulatory molecules upon programmed cell death.Citation133–135 These differences can be attributed to the origin of the tumor tissue,Citation136 underlying oncogenic events and (epi)genetic lesions the tumor has accumulated over time. Studies elucidating the direct effects of oncogenic events on the secretion and/or exposure of immunomodulatory molecules are generally lacking. However, a few recent studies have shown that oncogenesCitation137,Citation138 can influence the expression of immunomodulatory molecules during ICD. In the next sections, we review recent literature describing how oncogenic events can influence the level of immunomodulatory molecules emitted by malignant entities experiencing ICD ().
Calreticulin (CALR)
CALR is an endoplasmic reticulum resident protein critically important for maintaining calcium homeostasis and serves as a molecular chaperone to prevent the export of misfolded proteins to the Golgi apparatus.Citation139,Citation140 Calreticulin plays additional immunological functions such as facilitating the phagocytic uptake of dying tumor cells by innate immune cellsCitation129,Citation141 and serving as integral part of the peptide-loading complex for antigen presentation in the context of MHC-I.Citation142,Citation143 Moreover, CALR has many immune-related functions in T cells that have been extensively reviewed elsewhere.Citation93 Conversely, high expression of intracellular CALR promotes tumor cell proliferation contributing to metastasis in multiple cancer types.
Contrasting with the pro-tumor impact of intracellular CALR, surface-exposed CALR, following treatment with ICD-inducing chemotherapies, promotes tumor cell uptake by phagocytic cells.Citation129,Citation141 There are two ways by which CALR reaches the surface of dying cells. The first pathway relies on transcription and translational inhibition,Citation144 leading to the phosphorylation of eukaryotic initiation factor 2α (eIF2α) by the ER stress-sensing kinase, PKR-related ER kinase (PERK), and subsequent activation of caspase-8 and BAX. Finally, a pool of CALR that has transited the Golgi apparatus is secreted by SNAP receptor (SNARE)-dependent exocytosis.Citation145 In addition, paracrine signals that involve the chemokine CXCL8 contribute to CALR exposure in an eIF2a/PERK/BAX//BAK-dependent fashion.Citation146 Vaccination of mice with dying tumor cells deficient in any of the proteins required for CALR exposure, or with cells in which CALR was knocked down, reduced the immunogenicity of the dying cell vaccine.Citation145 The second pathway for CALR exposure is activated by photodynamic therapy and relies on direct PERK-mediated trafficking of CALR by regulation of the proximal secretory pathway.Citation147 In this signaling pathway, eIF2a phosphorylation and caspase-8 signaling are dispensable for CALR exposure.Citation147
Surface expression of CALR does not always occur after administration of cytotoxic therapies, as certain chemotherapies such as cisplatin or melphalan are unable to induce this feature of ICD.Citation148 Moreover, there are tumor-specific differences in surface exposure of CALR, as prototypical ICD inducers such as doxorubicin do not expose CALR on B-cell lymphomaCitation148 or acute myeloid leukemia cells.Citation149 Despite this, a retrospective analysis of non-small cell lung cancer patients showed that CALR expression on tumor cells was significantly correlated with eIF2α phosphorylation and disease outcome.Citation131 Higher CALR expression was associated with tumor infiltration by DCs and effector memory T-cell subsets that conferred prolonged survival.Citation131 Tumor-intrinsic determinants of CALR surface exposure are currently lacking. Recently, the promoter of the tumor suppressor retinoblastoma 1 (RB1) has been shown to dually control the expression of 7.1 kb non-coding RNA located upstream of the RB1 gene (ncRNA-RB1).Citation137 The study identified that CALR is a novel target of ncRNA-RB1. Depletion of ncRNA-RB1 contributes to a failed surface exposure of CALR during mitoxantrone treatment resulting in reduced phagocytosis of dying cells.Citation137 While surface CALR on dying cells promotes their phagocytic uptake, this process is often counteracted by tumors through overexpression of the anti-phagocytic signal CD47.151 CD47 is a pentaspanin cell surface protein that counters phagocytosis through ligation of its signal regulatory protein a (SIRPa) receptor on phagocytic cells.Citation150 Recently, oncogenic MYC was found to subvert immunosurveillance by upregulating the expression of CD47 which resulted in poor macrophage infiltration in multiple tumor types, including lymphoma/leukemia and liver cancerCitation138 ().
Extracellular ATP
Extracellular ATP actively secreted or passively released from dying tumors is one of the immunomodulatory molecules facilitating immune-mediated detection of tumors during ICD. The mechanism of ATP release during ICD depends on the type of lethal stimulus. Anthracyclines induce ATP release via caspase-dependent activation of pannexin 1 channels, lysosomal exocytosis, and plasma membrane blebbing.Citation151 Moreover, autophagy is indispensable for the release of ATP during anthracycline-induced tumor cell death. However, ATP secretion after hypericin-based PDT operates independently of autophagy and involves PERK-regulated proximal secretory pathways, as well as PI3K-dependent exocytosis. ATP exerts its immunomodulatory effect via activation of purinergic P2X7 and P2Y2 receptors (P2RX7, P2RY2) on myeloid cells. In mouse DCs, extracellular ATP binds to P2RX7 to initiate inflammasome signaling and the release of IL-1β to activate IL17-producing γδ T cells.Citation152 Thus, the lack of inflammasome signaling components and/or IL17 signaling aborts the immunogenic effects of anthracycline-induced cell death.Citation153
The excessive inflammatory effects of extracellular ATP are countered by a negative feedback mechanism to limit tissue injury.Citation154 Precisely, a system consisting of the ecto-enzymes CD39 and CD73, together with receptors of the adenosinergic pathway, converts immunostimulatory ATP into immunosuppressive adenosine.Citation155 ATP in the extracellular environment is broken down to adenosine monophosphate (AMP) by ecto-ATPases such as CD39.Citation155 The ecto-5ʹ-nucletodase CD73 breaks down AMP into adenosine. CD73-derived adenosine in the extracellular environment exerts its immunosuppressive effects by binding to one of the four G-protein-coupled adenosine receptors.Citation155 CD73 transcription is directly regulated by HIF-1alpha,Citation156 explaining why the hypoxic tumor microenvironment is often associated with high expression of CD73 in cancer cells, endothelial cells, fibroblasts, lymphocytes and myeloid cells. Tumor-derived CD73 can potently inhibit antitumor immunity by suppressing T and NK cell function.Citation157,Citation158 High amounts of CD73 in triple-negative breast cancerCitation132 and high-grade serous ovarian cancerCitation159 patients are negatively associated with tumor infiltration and overall patient survival. Patients with high levels of CD73 and low levels of tumor-infiltrating leucocytes show poor clinical outcome.Citation132 Oncogenic pathways such as Wnt and downstream β-catenin,Citation160 MAPK,Citation161 EGFRCitation162 and AKT signaling promote CD73 expression on tumor cellsCitation163 (). Moreover, increased CD73 was significantly associated with TP53 mutations in melanoma patients.Citation164 BRAF-targeted therapy with either dabrafenib or vermurafenib showed reduced levels of CD73 in biopsies of melanoma patients.Citation164
HMGB1
HMGB1 is a highly conserved nonhistone chromatic-binding protein abundantly expressed in all eukaryotic cells.Citation165,Citation166 HMGB1 can be released passively or actively,Citation167,Citation168 although the molecular mechanisms that initiate the release of HMGB1 from tumor cells undergoing ICD remain to be elucidated. Extracellular HMGB1 has several immunomodulatory propertiesCitation166,Citation169 based on its redox state and post-translational modifications.Citation170 The immunogenicity of anthracycline-treated dying apoptotic cells depends on the passive release of HMGB1 that binds to TLR4 in innate immune cells. Extracellular HMGB1 mediates its effects by binding to PRRs such as TLR4Citation171 and RAGE.Citation172 Various types of ICD inducers such as anthracyclines,Citation173 radiationCitation174–176 and oncolytic virusesCitation5 stimulate the secretion of HMGB1. The immunotherapeutic effects of anthracycline therapy failed in TLR4−/- and Myd88−/- mice suggesting that the HMGB1-TLR4-MYD88 axis is essential to stimulate the maturation of dendritic cells thereby enhancing their ability to cross-present tumor antigens.Citation173 In human breast cancer patients, a polymorphism in TLR4 that affects the binding of HMGB1 predicts relapse after anthracycline therapy.Citation173 In summary, the role of HMGB1 in anticancer immunity is complex and future studies should aim to elucidate tumor-specific and microenvironment-dependent immune effects of extracellular HMGB1.
Conclusions and future perspectives
The current state-of-the-art suggests that cancer cell-intrinsic factors can affect the activation and subversion of cancer cell ICD, although detailed mechanistic studies elucidating specific biological interactions are lacking. Understanding the intimate relationship between oncogenic factors and ICD will yield essential biological information on which oncogenic factors to target depending on the malignant indication. These investigations will be useful for the selection of patients that would benefit from immunomodulation upon ICD-inducing treatments. This knowledge will help maximizing the potential of ICD-inducing therapeutics for cancer patients and provide rationale for personalized medicine based on the genetic profile of tumors. We expect that the combination of targeted therapies against tumor-intrinsic oncogenic pathways with ICD-inducing agents will further stimulate the cancer-immunity cycle, and particularly the cytotoxic T cell response. This basic research will contribute to clinical oncology through the development of novel biotherapeutics.
Disclosure statement
JGP is named as an inventor on patents for cancer vaccination involving an oncolytic rhabdovirus. These patents have been licensed to Turnstone Biologics of which JGP is a shareholder. GK is a cofounder of Samsara Therapeutics, everImmune, and Therafast Bio. STW has no relevant conflict of interest to declare.
Additional information
Funding
References
- Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–13.
- Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. 2016;44(2):343–354.
- Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202(12):1691–1701.
- Lhuillier C, Rudqvist NP, Yamazaki T, Zhang T, Charpentier M, Galluzzi L, Dephoure N, Clement CC, Santambrogio L, Zhou XK, et al. Radiotherapy-exposed CD8+ and CD4+ neoantigens enhance tumor control. J Clin Invest. 2021.
- Workenhe ST, Simmons G, Pol JG, Lichty BD, Halford W, Mossman KL. Immunogenic HSV mediated oncolysis shapes the antitumor immune response and contributes to therapeutic efficacy. Mol Ther. 2014;22:123–131.
- Alzeibak R, Mishchenko TA, Shilyagina NY, Balalaeva IV, Vedunova MV, Krysko DV. Targeting immunogenic cancer cell death by photodynamic therapy: past, present and future. J Immunother Cancer. 2021;9(1).
- Coppard C, Hannani D, Humbert M, Gauthier V, Plumas J, Merlin E, Gabert F, Chaperot L. In vitro PUVA treatment triggers calreticulin exposition and HMGB1 release by dying T lymphocytes in GVHD: new insights in extracorporeal photopheresis. J Clin Apher. 2019;34:450–460.
- Tatsuno K, Yamazaki T, Hanlon D, Han P, Robinson E, Sobolev O, Yurter A, Rivera-Molina F, Arshad N, Edelson RL, et al. Extracorporeal photochemotherapy induces bona fide immunogenic cell death. Cell Death Dis. 2019;10(8):578.
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723.
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133.
- Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Hegg R, Im S-A, Shaw Wright G, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379(22):2108–2121.
- Lévesque S, Le Naour J, Pietrocola F, Paillet J, Kremer M, Castoldi F, Baracco EE, Wang Y, Vacchelli E, Stoll G, et al. A synergistic triad of chemotherapy, immune checkpoint inhibitors, and caloric restriction mimetics eradicates tumors in mice. Oncoimmunology. 2019;8(11):e1657375.
- Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217–223.
- Najafov A, Zervantonakis IK, Mookhtiar AK, Greninger P, March RJ, Egan RK, Luu HS, Stover DG, Matulonis UA, Benes CH, et al. BRAF and AXL oncogenes drive RIPK3 expression loss in cancer. PLoS Biol. 2018;16(8):e2005756.
- Sallusto F, Cella M, Danieli C, Lanzavecchia A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J Exp Med. 1995;182:389–400.
- Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, et al. Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. 2016;44(4):924–938.
- Han P, Hanlon D, Arshad N, Lee JS, Tatsuno K, Robinson E, Filler R, Sobolev O, Cote C, Rivera-Molina F, et al. Platelet P-selectin initiates cross-presentation and dendritic cell differentiation in blood monocytes. Sci Adv. 2020;6(11):eaaz1580.
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10.
- Pol JG, Caudana P, Paillet J, Piaggio E, Kroemer G. Effects of interleukin-2 in immunostimulation and immunosuppression. J Exp Med. 2020;217(1).
- Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front Immunol. 2018;9:847.
- Montfort A, Colacios C, Levade T, Andrieu-Abadie N, Meyer N, Ségui B. The TNF paradox in cancer progression and immunotherapy. Front Immunol. 2019;10:1818.
- Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, McSkane M, Baba H, Lenz HJ. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - A target for novel cancer therapy. Cancer Treat Rev. 2018;63:40–47.
- Kägi D, Ledermann B, Bürki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, Hengartner H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369:31–37.
- Van Den Broek ME, Kägi D, Ossendorp F, Toes R, Vamvakas S, Lutz WK, Melief CJ, Zinkernagel RM, Hengartner H. Decreased tumor surveillance in perforin-deficient mice. J Exp Med. 1996;184:1781–1790.
- Ghorani E, Reading JL, Henry JY, Massy M, Rosenthal R, Turati V, Joshi K, Furness AJS, Ben AA, Saini SK, et al. The T cell differentiation landscape is shaped by tumour mutations in lung cancer. Nature Cancer. 2020;1(5):546–561.
- Ozcan M, Janikovits J, Von Knebel Doeberitz M, Kloor M. Complex pattern of immune evasion in MSI colorectal cancer. Oncoimmunology. 2018;7:e1445453.
- Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61.
- Chen S, Crabill GA, Pritchard TS, McMiller TL, Wei P, Pardoll DM, Pan F, Topalian SL. Mechanisms regulating PD-L1 expression on tumor and immune cells. J Immunother Cancer. 2019;7:305.
- Labadie BW, Bao R, Luke JJ. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan-kynurenine-aryl hydrocarbon axis. Clin Cancer Res. 2019;25:1462–1471.
- Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19(6):1189–1201.
- Moon JW, Kong SK, Kim BS, Kim HJ, Lim H, Noh K, Kim Y, Choi JW, Lee JH, Kim YS. IFNγ induces PD-L1 overexpression by JAK2/STAT1/IRF-1 signaling in EBV-positive gastric carcinoma. Sci Rep. 2017;7:17810.
- Yang B, Liu T, Qu Y, Liu H, Zheng SG, Cheng B, Sun J. Progresses and perspectives of anti-PD-1/PD-L1 antibody therapy in head and neck cancers. Front Oncol. 2018;8:563.
- Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, Fatho M, Lennerz V, Wölfel T, Hölzel M, et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature. 2012;490(7420):412–416.
- Ribas A, Tumeh PC. Cancer therapy: tumours switch to resist. Nature. 2012;490:347–348.
- Hoek KS, Eichhoff OM, Schlegel NC, Döbbeling U, Kobert N, Schaerer L, Hemmi S, Dummer R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008;68:650–656.
- Hölzel M, Bovier A, Tüting T. Plasticity of tumour and immune cells: a source of heterogeneity and a cause for therapy resistance? Nat Rev Cancer. 2013;13:365–376.
- Ribas A. Adaptive immune resistance: how cancer protects from immune attack. Cancer Discov. 2015;5:915–919.
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW. Cancer genome landscapes. Science (New York, NY). 2013;339:1546–1558.
- Reiter JG, Baretti M, Gerold JM, Makohon-Moore AP, Daud A, Iacobuzio-Donahue CA, Azad NS, Kinzler KW, Nowak MA, Vogelstein B. An analysis of genetic heterogeneity in untreated cancers. Nat Rev Cancer. 2019;19:639–650.
- Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6:715–727.
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–829.
- Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, Richman LP, Lin JH, Sun YH, Rech AJ, et al. Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity. 2018;49(1):178–93.e7.
- Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938.
- Wellenstein MD, De Visser KE. Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity. 2018;48:399–416.
- Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. 2011;42:23–35.
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602.
- Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837.
- Han C, Liu Z, Zhang Y, Shen A, Dong C, Zhang A, Moore C, Ren Z, Lu C, Cao X, et al. Tumor cells suppress radiation-induced immunity by hijacking caspase 9 signaling. Nat Immunol. 2020;21(5):546–554.
- Ning X, Wang Y, Jing M, Sha M, Lv M, Gao P, Zhang R, Huang X, Feng JM, Jiang Z. Apoptotic caspases suppress type i interferon production via the cleavage of cGAS, MAVS, and IRF3. Mol Cell. 2019;74:19–31.e7.
- Rodriguez-Ruiz ME, Buqué A, Hensler M, Chen J, Bloy N, Petroni G, Sato A, Yamazaki T, Fucikova J, Galluzzi L. Apoptotic caspases inhibit abscopal responses to radiation and identify a new prognostic biomarker for breast cancer patients. Oncoimmunology. 2019;8:e1655964.
- Hanahan D, Weinberg Robert A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674.
- Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111.
- Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer. 2002;2:277–288.
- Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 2016;14:282–297.
- Shi S, Xu C, Fang X, Zhang Y, Li H, Wen W, Yang G. Expression profile of Tolllike receptors in human breast cancer. Mol Med Rep. 2020;21:786–794.
- Zhu H, Xu W-Y, Hu Z, Zhang H, Shen Y, Lu S, Wei C, Wang Z-G. RNA virus receptor Rig-I monitors gut microbiota and inhibits colitis-associated colorectal cancer. J Exp Clin Cancer Res. 2017;36:2.
- Ding HF, McGill G, Rowan S, Schmaltz C, Shimamura A, Fisher DE. Oncogene-dependent regulation of caspase activation by p53 protein in a cell-free system. J Biol Chem. 1998;273:28378–28383.
- Amarante-Mendes GP, Finucane DM, Martin SJ, Cotter TG, Salvesen GS, Green DR. Anti-apoptotic oncogenes prevent caspase-dependent and independent commitment for cell death. Cell Death Differ. 1998;5:298–306.
- Towers CG, Wodetzki D, Thorburn A. Autophagy and cancer: modulation of cell death pathways and cancer cell adaptations. J Cell Biol. 2019;jcb.201909033.
- Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34(7):856–880.
- Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, Tavaré S, Arakawa S, Shimizu S, Watt FM, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23(7):798–803.
- Liu H, He Z, Von Rütte T, Yousefi S, Hunger RE, Simon H-U. Down-regulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci Transl Med. 2013;5:202ra123–202ra123.
- Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science (New York, NY). 2011;333:1109–1112.
- Takahashi Y, Hori T, Cooper TK, Liao J, Desai N, Serfass JM, Young MM, Park S, Izu Y, Wang H-G. Bif-1 haploinsufficiency promotes chromosomal instability and accelerates Myc-driven lymphomagenesis via suppression of mitophagy. Blood. 2013;121:1622–1632.
- Rodriguez OC, Choudhury S, Kolukula V, Vietsch EE, Catania J, Preet A, Reynoso K, Bargonetti J, Wellstein A, Albanese C, et al. Dietary downregulation of mutant p53 levels via glucose restriction: mechanisms and implications for tumor therapy. Cell Cycle (Georgetown, Tex). 2012;11(23):4436–4446.
- Choudhury S, Kolukula VK, Preet A, Albanese C, Avantaggiati ML. Dissecting the pathways that destabilize mutant p53: the proteasome or autophagy? Cell Cycle (Georgetown, Tex). 2013;12:1022–1029.
- Isakson P, Bjørås M, Bøe SO, Simonsen A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood. 2010;116:2324–2331.
- Goussetis DJ, Gounaris E, Wu EJ, Vakana E, Sharma B, Bogyo M, Altman JK, Platanias LC. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood. 2012;120:3555–3562.
- Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJR, Motoyama N, Cao L, Finkel T. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science (New York, NY). 2012;336:225–228.
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676.
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–1820.
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–15082.
- Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, Stranks AJ, Glanville J, Knight S, Jacobsen S-EW, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011;208(3):455–467.
- Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, McMahon M, White E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013;3:1272–1285.
- Guo JY, Teng X, Laddha SV, Ma S, Van Nostrand SC, Yang Y, Khor S, Chan CS, Rabinowitz JD, White E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016;30:1704–1717.
- Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4(8):914–927.
- Katheder NS, Khezri R, O’Farrell F, Schultz SW, Jain A, Rahman MM, Schink KO, Theodossiou TA, Johansen T, Juhász G, et al. Microenvironmental autophagy promotes tumour growth. Nature. 2017;541(7637):417–420.
- Rao S, Yang H, Penninger JM, Kroemer G. Autophagy in non-small cell lung carcinogenesis: a positive regulator of antitumor immunosurveillance. Autophagy. 2014;10:529–531.
- Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, Von Hoff DD, Maitra A, Kimmelman AC. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014;4:905–913.
- Lazova R, Camp RL, Klump V, Siddiqui SF, Amaravadi RK, Pawelek JM. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin Cancer Res. 2012;18:370–379.
- Pietrocola F, Pol J, Kroemer G. Fasting improves anticancer immunosurveillance via autophagy induction in malignant cells. Cell Cycle. 2016;15:3327–3328.
- Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, Portela Catani JP, Hannani D, Duret H, Steegh K, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38(4):729–741.
- Michaud M, Xie X, Bravo-San Pedro JM, Zitvogel L, White E, Kroemer G. An autophagy-dependent anticancer immune response determines the efficacy of melanoma chemotherapy. Oncoimmunology. 2014;3:e944047.
- Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, Levesque S, Castoldi F, Jacquelot N, Yamazaki T, et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell. 2016;30(1):147–160.
- Uhl M, Kepp O, Jusforgues-Saklani H, Vicencio JM, Kroemer G, Albert ML. Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8+ T cells. Cell Death Differ. 2009;16:991–1005.
- Xu X, Araki K, Li S, Han J-H, Ye L, Tan WG, Konieczny BT, Bruinsma MW, Martinez J, Pearce EL, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol. 2014;15(12):1152–1161.
- Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, Van Moer K, Kreis S, Guerin C, Buart S, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proceedings of the National Academy of Sciences of the United States of America 2017;114( 44):E9271–E9.
- Maiuri MC, Tasdemir E, Criollo A, Morselli E, Vicencio JM, Carnuccio R, Kroemer G. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009;16:87–93.
- Morselli E, Galluzzi L, Kepp O, Mariño G, Michaud M, Vitale I, Maiuri MC, Kroemer G. Oncosuppressive functions of autophagy. Antioxid Redox Signal. 2011;14:2251–2269.
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939.
- Maiuri MC, Le Toumelin G, Criollo A, Rain J-C, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26(10):2527–2539.
- Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15:1126–1132.
- Furuta S, Hidaka E, Ogata A, Yokota S, Kamata T. Ras is involved in the negative control of autophagy through the class I PI3-kinase. Oncogene. 2004;23:3898–3904.
- Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320.
- Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, Avanzi A, Barilla R, Daley D, Greco SH, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 2016;532(7598):245–249.
- Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira Da Silva R, Reis E Sousa C, Green DR, Oberst A, Albert ML. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science. 2015;350:328–334.
- Van Hoecke L, Van Lint S, Roose K, Van Parys A, Vandenabeele P, Grooten J, Tavernier J, De Koker S, Saelens X. Treatment with mRNA coding for the necroptosis mediator MLKL induces antitumor immunity directed against neo-epitopes. Nat Commun. 2018;9:3417.
- Snyder AG, Hubbard NW, Messmer MN, Kofman SB, Hagan CE, Orozco SL, Chiang K, Daniels BP, Baker D, Oberst A. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci Immunol. 2019;4:eaaw2004.
- Wang W, Marinis JM, Beal AM, Savadkar S, Wu Y, Khan M, Taunk PS, Wu N, Su W, Wu J, et al. RIP1 kinase drives macrophage-mediated adaptive immune tolerance in pancreatic cancer. Cancer Cell. 2018;34(5):757–74 e7.
- Koo G-B, Morgan MJ, Lee D-G, Kim W-J, Yoon J-H, Koo JS, Kim SI, Kim SJ, Son MK, Hong SS, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015;25(6):707–725.
- Fukasawa M, Kimura M, Morita S, Matsubara K, Yamanaka S, Endo C, Sakurada A, Sato M, Kondo T, Horii A, et al. Microarray analysis of promoter methylation in lung cancers. J Hum Genet. 2006;51(4):368–374.
- Nugues AL, El Bouazzati H, Hétuin D, Berthon C, Loyens A, Bertrand E, Jouy N, Idziorek T, Quesnel B. RIP3 is downregulated in human myeloid leukemia cells and modulates apoptosis and caspase-mediated p65/RelA cleavage. Cell Death Dis. 2014;5:e1384–e.
- Moriwaki K, Bertin J, Gough PJ, Orlowski GM, Chan FK. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015;6:e1636–e.
- Lalaoui N, Brumatti G. Relevance of necroptosis in cancer. Immunol Cell Biol. 2017;95:137–145.
- Zhang YY, Tabataba H, Liu XY, Wang JY, Yan XG, Farrelly M, Jiang CC, Guo ST, Liu T, Kao H-Y, et al. ACTN4 regulates the stability of RIPK1 in melanoma. Oncogene. 2018;37(29):4033–4045.
- Park S, Hatanpaa KJ, Xie Y, Mickey BE, Madden CJ, Raisanen JM, Ramnarain DB, Xiao G, Saha D, Boothman DA, et al. The receptor interacting protein 1 Inhibits p53 Induction through NF-κB activation and confers a worse prognosis in glioblastoma. Cancer Res. 2009;69(7):2809–2816.
- Wang Q, Chen W, Xu X, Li B, He W, Padilla MT, Jang J-H, Nyunoya T, Amin S, Wang X, et al. RIP1 potentiates BPDE-induced transformation in human bronchial epithelial cells through catalase-mediated suppression of excessive reactive oxygen species. Carcinogenesis. 2013;34(9):2119–2128.
- McCabe KE, Bacos K, Lu D, Delaney JR, Axelrod J, Potter MD, Vamos M, Wong V, Cosford NDP, Xiang R, et al. Triggering necroptosis in cisplatin and IAP antagonist-resistant ovarian carcinoma. Cell Death Dis. 2014;5(10):e1496–e.
- Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8:1812–1825.
- Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99.
- Rogers C, Alnemri ES. Gasdermins: novel mitochondrial pore-forming proteins. Mol Cell Oncol. 2019;6:e1621501.
- Karki R, Kanneganti T-D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat Rev Cancer. 2019;19:197–214.
- Daley D, Mani VR, Mohan N, Akkad N, Pandian GSDB, Savadkar S, Lee KB, Torres-Hernandez A, Aykut B, Diskin B, et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J Exp Med. 2017;214(6):1711.
- Saijo Y, Tanaka M, Miki M, Usui K, Suzuki T, Maemondo M, Hong X, Tazawa R, Kikuchi T, Matsushima K, et al. Proinflammatory cytokine IL-1 beta promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors: in vivo analysis of tumor-stromal interaction. Jl Immunol. 2002;169(1):469–475. Baltimore, Md: 1950.
- Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, Betz KS, Penz-Oesterreicher M, Bjorkdahl O, Fox JG, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14(5):408–419.
- Nakamura K, Kassem S, Cleynen A, Chrétien M-L, Guillerey C, Putz EM, Bald T, Förster I, Vuckovic S, Hill GR, et al. Dysregulated IL-18 is a key driver of immunosuppression and a possible therapeutic target in the multiple myeloma microenvironment. Cancer Cell. 2018;33(4):634–48.e5.
- Terme M, Ullrich E, Aymeric L, Meinhardt K, Desbois M, Delahaye N, Viaud S, Ryffel B, Yagita H, Kaplanski G, et al. IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer Res. 2011;71(16):5393–5399.
- Kang JS, Bae SY, Kim HR, Kim YS, Kim DJ, Cho BJ, Yang H-K, Hwang Y-I, Kim KJ, Park HS, et al. Interleukin-18 increases metastasis and immune escape of stomach cancer via the downregulation of CD70 and maintenance of CD44. Carcinogenesis. 2009;30(12):1987–1996.
- Williams TM, Leeth RA, Rothschild DE, Coutermarsh-Ott SL, McDaniel DK, Simmons AE, Heid B, Cecere TE, Allen IC. The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. Jl Immunol. 2015;194(7):3369–3380. Baltimore, Md: 1950.
- Allen IC, TeKippe EM, Woodford R-MT, Uronis JM, Holl EK, Rogers AB, Herfarth HH, Jobin C, Ting JPY. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med. 2010;207:1045–1056.
- Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, Kanneganti T-D. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. Jl Immunol. 2010;185(8):4912–4920. Baltimore, Md: 1950.
- Wilson JE, Petrucelli AS, Chen L, Koblansky AA, Truax AD, Oyama Y, Rogers AB, Brickey WJ, Wang Y, Schneider M, et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat Med. 2015;21(8):906–913.
- Kim MS, Lebron C, Nagpal JK, Chae YK, Chang X, Huang Y, Chuang T, Yamashita K, Trink B, Ratovitski EA, et al. Methylation of the DFNA5 increases risk of lymph node metastasis in human breast cancer. Biochem Biophys Res Commun. 2008;370(1):38–43.
- Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, Wang Y, Li D, Liu W, Zhang Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020.
- Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, Junqueira C, Meza-Sosa KF, Mok TMY, Ansara J, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579(7799):415–420.
- Akino K, Toyota M, Suzuki H, Imai T, Maruyama R, Kusano M, Nishikawa N, Watanabe Y, Sasaki Y, Abe T, et al. Identification of DFNA5 as a target of epigenetic inactivation in gastric cancer. Cancer Sci. 2007;98(1):88–95.
- Ibrahim J, Op De Beeck K, Fransen E, Croes L, Beyens M, Suls A, Vanden BW, Peeters M, Van CG. Methylation analysis of Gasdermin E shows great promise as a biomarker for colorectal cancer. Cancer Med. 2019;8:2133–2145.
- Lu H, Zhang S, Wu J, Chen M, Cai M-C, Fu Y, Li W, Wang J, Zhao X, Yu Z, et al. Molecular targeted therapies elicit concurrent apoptotic and GSDME-dependent pyroptotic tumor cell death. Clin Cancer Res. 2018;24(23):6066–6077.
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54–61.
- Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334(6062):1573–1577.
- Fucikova J, Becht E, Iribarren K, Goc J, Remark R, Damotte D, Alifano M, Devi P, Biton J, Germain C, et al. Calreticulin expression in human non–small cell lung cancers correlates with increased accumulation of antitumor immune cells and favorable prognosis. Cancer Res. 2016;76(7):1746–1756.
- Buisseret L, Pommey S, Allard B, Garaud S, Bergeron M, Cousineau I, Ameye L, Bareche Y, Paesmans M, Crown JPA, et al. Clinical significance of CD73 in triple-negative breast cancer: multiplex analysis of a phase III clinical trial. Ann Oncol. 2017;29(4):1056–1062.
- Coffelt SB, De Visser KE. Immune-mediated mechanisms influencing the efficacy of anticancer therapies. Trends Immunol. 2015;36:198–216.
- Zitvogel L, Kroemer G. Reply to: chemotherapy response of spontaneous mammary tumors is independent of the adaptive immune system. Nat Med. 2012;18:346.
- Garg AD, Agostinis P. Editorial: immunogenic cell death in cancer: from benchside research to bedside reality. Front Immunol. 2016;7:110.
- Salmon H, Remark R, Gnjatic S, Merad M. Host tissue determinants of tumour immunity. Nat Rev Cancer. 2019;19:215–227.
- Musahl AS, Huang X, Rusakiewicz S, Ntini E, Marsico A, Kroemer G, Kepp O, Ørom UA. A long non-coding RNA links calreticulin-mediated immunogenic cell removal to RB1 transcription. Oncogene. 2015;34:5046–5054.
- Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gütgemann I, Eilers M, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science (New York, NY). 2016;352(6282):227–231.
- Eggleton P, Bremer E, Dudek E, Michalak M. Calreticulin, a therapeutic target? Expert Opin Ther Targets. 2016;20:1137–1147.
- Michalak M, Corbett EF, Mesaeli N, Nakamura K, Opas M. Calreticulin: one protein, one gene, many functions. Biochem J. 1999;344:281–292.
- Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg P-A, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–334.
- Sadasivan B, Lehner PJ, Ortmann B, Spies T, Cresswell P. Roles for calreticulin and a novel glycoprotein, tapasin, in the interaction of MHC class I molecules with TAP. Immunity. 1996;5:103–114.
- Peaper DR, Cresswell P. Regulation of MHC class I assembly and peptide binding. Annu Rev Cell Dev Biol. 2008;24:343–368.
- Humeau J, Sauvat A, Cerrato G, Xie W, Loos F, Iannantuoni F, Bezu L, Lévesque S, Paillet J, Pol J, et al. Inhibition of transcription by dactinomycin reveals a new characteristic of immunogenic cell stress. EMBO Mol Med. 2020;12(5):e11622.
- Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund A-C, Chapman DC, Durchschlag M, Joza N, Pierron G, Van Endert P, et al. Mechanisms of pro-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28(5):578–590.
- Sukkurwala AQ, Martins I, Wang Y, Schlemmer F, Ruckenstuhl C, Durchschlag M, Michaud M, Senovilla L, Sistigu A, Ma Y, et al. Immunogenic calreticulin exposure occurs through a phylogenetically conserved stress pathway involving the chemokine CXCL8. Cell Death Differ. 2014;21(1):59–68.
- Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJM, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012;31(5):1062–1079.
- Kang JS, Dervan PB. A sequence-specific DNA binding small molecule triggers the release of immunogenic signals and phagocytosis in a model of B-cell lymphoma. Q Rev Biophys. 2015;48:453–464.
- Wemeau M, Kepp O, Tesnière A, Panaretakis T, Flament C, De Botton S, Zitvogel L, Kroemer G, Chaput N. Calreticulin exposure on malignant blasts predicts a cellular anticancer immune response in patients with acute myeloid leukemia. Cell Death Dis. 2010;1:e104–e.
- Liu X, Kwon H, Li Z, Fu Y-X. Is CD47 an innate immune checkpoint for tumor evasion? J Hematol Oncol. 2017;10:12.
- Sandilos JK, Chiu Y-H, Chekeni FB, Armstrong AJ, Walk SF, Ravichandran KS, Bayliss DA. Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J Biol Chem. 2012;287:11303–11311.
- Ma Y, Aymeric L, Locher C, Mattarollo SR, Delahaye NF, Pereira P, Boucontet L, Apetoh L, Ghiringhelli F, Casares N, et al. Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J Exp Med. 2011;208(3):491–503.
- Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15(10):1170–1178.
- Allard D, Chrobak P, Allard B, Messaoudi N, Stagg J. Targeting the CD73-adenosine axis in immuno-oncology. Immunol Lett. 2019;205:31–39.
- Robson SC, Sévigny J, Zimmermann H. The E-NTPDase family of ectonucleotidases: structure function relationships and pathophysiological significance. Purinergic Signal. 2006;2:409–430.
- Sitkovsky MV, Hatfield S, Abbott R, Belikoff B, Lukashev D, Ohta A. Hostile, hypoxia-A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol Res. 2014;2:598–605.
- Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, Dwyer KM, Smyth MJ Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proceedings of the National Academy of Sciences of the United States of America 2010;107( 4):1547–1552.
- Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ, Shin T, Curiel TJ, Zhang B. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res. 2010;70:2245–2255.
- Turcotte M, Spring K, Pommey S, Chouinard G, Cousineau I, George J, Chen GM, Gendoo DMA, Haibe-Kains B, Karn T, et al. CD73 is associated with poor prognosis in high-grade serous ovarian cancer. Cancer Res. 2015;75(21):4494–4503.
- Spychala J, Kitajewski J. Wnt and beta-catenin signaling target the expression of ecto-5ʹ-nucleotidase and increase extracellular adenosine generation. Exp Cell Res. 2004;296:99–108.
- Reinhardt J, Landsberg J, Schmid-Burgk JL, Ramis BB, Bald T, Glodde N, Lopez-Ramos D, Young A, Ngiow SF, Nettersheim D, et al. MAPK signaling and inflammation link melanoma phenotype switching to induction of CD73 during immunotherapy. Cancer Res. 2017;77(17):4697–4709.
- Inoue Y, Yoshimura K, Kurabe N, Kahyo T, Kawase A, Tanahashi M, Ogawa H, Inui N, Funai K, Shinmura K, et al. Prognostic impact of CD73 and A2A adenosine receptor expression in non-small-cell lung cancer. Oncotarget. 2017;8(5):8738–8751.
- Zhu J, Zeng Y, Li W, Qin H, Lei Z, Shen D, Gu D, Huang J-A, Liu Z. CD73/NT5E is a target of miR-30a-5p and plays an important role in the pathogenesis of non-small cell lung cancer. Mol Cancer. 2017;16:34.
- Young A, Ngiow SF, Madore J, Reinhardt J, Landsberg J, Chitsazan A, Rautela J, Bald T, Barkauskas DS, Ahern E, et al. Targeting adenosine in BRaf-mutant melanoma reduces tumor growth and metastasis. Cancer Res. 2017;77(17):4684–4696.
- Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388.
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195.
- Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560.
- Lu B, Antoine DJ, Kwan K, Lundbäck P, Wähämaa H, Schierbeck H, Robinson M, Van Zoelen MAD, Yang H, Li J, et al. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proceedings of the National Academy of Sciences 2014;111( 8):3068–3073.
- Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285(5425):248–251.
- Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A, Raeli L, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209(9):1519–1528.
- Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proceedings of the National Academy of Sciences 2010;107( 26):11942–11947.
- LeBlanc PM, Doggett TA, Choi J, Hancock MA, Durocher Y, Frank F, Nagar B, Ferguson TA, Saleh M. An immunogenic peptide in the A-box of HMGB1 protein reverses apoptosis-induced tolerance through RAGE receptor. J Biol Chem. 2014;289:7777–7786.
- Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, Mariette C, Chaput N, Mira JP, Delaloge S, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007;220:47–59.
- Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology. 2014;3:e28518–e.
- Brusa D, Migliore E, Garetto S, Simone M, Matera L. Immunogenicity of 56 degrees C and UVC-treated prostate cancer is associated with release of HSP70 and HMGB1 from necrotic cells. Prostate. 2009;69:1343–1352.
- Werthmöller N, Frey B, Wunderlich R, Fietkau R, Gaipl US. Modulation of radiochemoimmunotherapy-induced B16 melanoma cell death by the pan-caspase inhibitor zVAD-fmk induces anti-tumor immunity in a HMGB1-, nucleotide- and T-cell-dependent manner. Cell Death Dis. 2015;6:e1761–e.