
ABSTRACT
The role of T cells in controlling human cancers is well known. Their success requires continued persistence in vivo and efficient trafficking to tumor sites, requirements shared by other effectors such as Natural Killer (NK) cells. To date, cytokine IL2 remains the only clinically approved cytokine therapy available to expand, maintain, and activate these effector lymphoid cells, but toxicities can be severe. Cytokine IL15 offers similar T cell proliferation and activation properties, but without the unwanted side-effects seen with IL2. Several IL15-cytokine fusion proteins have been developed to improve their in vivo function, typically exploiting the IL15Rα to complex with IL15, to extend serum half-life and increase affinity for IL15β receptor on immune cells. Here we describe a novel IL15 complex incorporating the full-length IL15Rα to complex with wild type IL15 to form spontaneous trimers of dimers (6 IL15 + 6 IL15Rα) during co-expression, resulting in a substantial increase in serum half-life and enhancement of in vivo cytokine effect on IgG or T cell engaging antibody-dependent cell-mediated cytotoxicities, when compared to alternative strategies.
Introduction
Despite the enormous advances in the therapeutic application of innate and adaptive immunities to human cancer, the hurdles preventing effective anti-tumor responses in solid tumors remain daunting.Citation1 Tumors like neuroblastoma downregulate their class I and class II HLA antigens to evade the afferent and efferent arms of adaptive immunity,Citation2 partly explaining the paucity of tumor-infiltrating lymphocytes (TILs) in these “cold” tumors.Citation3 For neuroblastoma, innate immunity is the only clinically approved anti-tumor treatment so far, where anti-disialoganglioside GD2 monoclonal antibodies successfully exploit immune effector cells expressing Fc-receptors.Citation4,Citation5
Recently, T cells have emerged as one of the most promising approaches to eradicate certain types of leukemia and solid tumors. Immune checkpoint blockade therapies have assumed center stage, where removing the “brakes” in T cells has achieved unprecedented clinical responses among patients with metastatic cancers.Citation6,Citation7 Such immune checkpoint blockade antibodies can also play a role in natural killer (NK) cell-mediated anti-tumor cytotoxicity.Citation8 However, one major limit to these successes is the insufficient preexisting immunity and tumor-infiltrating lymphocytes (TILs). Since tumor-specific T cells are rare in patients with high-risk neuroblastoma,Citation3 chimeric antigen receptors (anti-GD2) have been developed to “educate” T cells to recognize tumor surface antigens,Citation9 although their persistence and exhaustion in vivo after adoptive transfer are recurrent hurdles. Another strategy currently in clinical development uses bispecific antibodies (BsAb) to redirect polyclonal T cells to achieve durable tumor response (NCT03860207).Citation10 However, other than IL2, there is no clinically available pharmaceuticals that will expand, maintain and activate these redirected T cells.
Cytokine interleukin-15 (IL15) has diverse immunologic effects, and plays an important role in the development, homeostasis and function of memory CD8+ T cells, NK cells, and other immune cells (reviewed inCitation11). IL15 is a 14–15 kDa glycoprotein that binds to a heterotrimeric receptor, composed of a unique alpha subunit (IL15Rα) that confers receptor specificity, and the IL2R/IL15Rβ (CD122) and the common gamma (γc) chain (CD132) shared with the IL2 receptor.Citation12 However, unlike IL2, IL15 does not (i) activate T regulatory cells (Tregs),Citation13 (ii) induce activation-induced cell death (AICD) among CD8(+)T cells,Citation14 (iii) cause vascular capillary leak,Citation15 and (iv) IL15 can sustain the survival of NK, effector CD8(+) and memory phenotype CD8(+) T cells. IL15Rα contains three protein domains: (i) a 175 amino acid extracellular domain, (ii) a single 23 amino acid transmembrane region, and (iii) a 39 amino acid cytoplasmic domain, and is widely expressed in humans and mice independently of IL2R/IL15Rβγc.Citation14 IL15Ra binds to IL15 with extremely high affinity (Kd <10–11 M) allowing it to retain IL15 on the cell surface. Through this, IL15Rα can trans-present IL15 to IL2R/IL15Rβγc on neighboring NK and T cells through immunological synapses.Citation16 This synapse mechanism limits exposure to circulating IL15, thereby decreasing the risk of autoimmunity. IL15 can also persist for days in a membrane-bound form by recycling as complex with IL15Rα through endosomal vesicles.Citation16 IL15 was ranked 1st among a list of agents with high potential to serve as immunotherapeutic drugs by the NCI immunotherapy agent workshop.Citation17
One common strategy to extend serum half-life uses Fc-fusion proteins based on IgG Fc domains, resulting in usually dimeric proteins with MW between 100 and 160 kDa. This half-life extension has been proven in many Fc fusion systems, with a TNF-α receptor Fc-fusion extending serum half-life 5-fold, and improving antagonistic potencies 1000-fold.Citation18 Previous attempts to develop IL15-Fc-Fusion proteins have exploited the minimal IL15 binding domain of the IL15Ra, named the sushi-binding domain (aa 1–66), to improve half-life and potency.Citation19 More recently, others have modified the IL15 sequence itself, mutating a single amino acid (N72D) to increase binding to IL15Rβ, and combining this in an Fc-fusion complex with an IL15Ra sushi domain (ALT-803), showing potent pre-clinical activity to warrant patient trials.Citation19,Citation20 Besides, full-length IL15Ra-IgG1-Fc fusion protein has been shown to enhance anti-tumor activity through NK and CD8+ T Cells proliferation and activation when premixed with IL15.Citation21
In this report, we describe a novel IL15/IL15Ra Fc fusion protein (WT-FL) using the full-length IL15Rα that multimerizes into a trimers of dimers (6 IL15 + 6 IL15Rα) during co-expression, resulting in substantial increases in serum half-life and in vivo anti-tumor efficacy, compared to fusion proteins using the truncated sushi domain. This WT-FL protein synergizes with both IgG and BsAb therapies in vivo, using either xenograft or syngeneic tumor models, as proof of principle for its potential combination with antibodies in clinical development.
Materials and Methods
Cell lines
Human neuroblastoma cell line IMR32 and mouse lymphoblast cell line CTLL-2 were purchased from ATCC (Manassas, VA). Human melanoma cell line M14 was obtained from UCLA, and murine melanoma cell line B78/D14 from Dr. Kenneth Lloyd, MSK.Citation22 All cells were authenticated by short tandem repeat profiling using PowerPlex 1.2 System (Promega), and periodically tested for mycoplasma using a commercial kit (Lonza). The luciferase-labeled tumor cell lines IMR32-Luc and M14-Luc were generated by retroviral infection with an SFG-GFLuc vector.
Cloning, co-expression and purification of IL15/IL15Rα-Fc complexes
Full-length ectodomain of IL15Rα (aa1-175) was fused to the CH2-CH3 of human IgG1 Fc region. Wild type (WT) and N72D mutated IL15 (MUT), in complex with either full length (FL) or Sushi domain (SU, aa1-66) of IL15Rα were made for comparison. These genes were synthesized by Genscript with appropriate flanking restriction enzyme sites, subcloned into a single two-segment mammalian expression vector, and used to transfect CHO-S cells (Invitrogen) for stable co-expression of protein complex and selection of high producers. 1 × 106 cells were transfected with 2.5 µg of linearized plasmid DNA by Nucleofection (Lonza) and then recovered in CD OptiCHO media supplemented with 8 mM L-glutamine (Invitrogen) for 2 days at 37°C in 6-well culture plate. Stable pools were selected first with 500 ug/ml hygromycin for approximately 2 weeks, single clones were then selected out with limited dilution. IL15/IL15Rα-Fc titer was determined by ELISA, where plates were coated with anti-human IL15 polyclonal goat IgG (R&D Systems) to capture the complex, and then detected with secondary goat anti-human IgG (Fc specific) (Southern Biotech). High expression clones with strong binding signals were selected for further subcloning. The complexes were purified using Protein A affinity chromatography.
Biochemical characterization of IL15/IL15Rα-Fc complexes
Ten micrograms of the protein was analyzed by SDS-PAGE using 4–15% Tris-Glycine Ready Gel System (Bio-Rad). Invitrogen SeeBlue Plus2 Pre-Stained Standard, or Life Technologies BenchMark Unstained Protein Ladder was used as the protein MW marker. After electrophoresis, the gel was stained using Coomassie G-250 (GelCode Blue Stain Reagent; Pierce). The size and purity of complexes was also evaluated by size-exclusion high-performance liquid chromatography (SE-HPLC). Approximately 20 µg of protein was injected into a TSK-GEL G4000SWXL 7.8 mm x 30 cm, 8 µm column (TOSOH Bioscience) with 0.4 M NaClO4, 0.05 M NaH2PO4, pH 6.0 buffer at flow rate of 0.5 mL/min, and UV detection at 280 nm. Ten microliters of gel-filtration standard (Sigma) was analyzed in parallel for MW markers.
In vitro cell proliferation assays
For cell proliferation, mouse lymphoblast CTLL-2 cells (15,000 cells/well) plated using RPMI-1640 supplemented with 10% FBS in a 96 well plate were incubated with IL15 complexes at the specified concentration for 72 hrs at 37°C. Cell proliferation was determined using the cell counting WST-8 kit (Dojindo Molecular Technologies) following the manufacturer’s instructions.
Cytotoxicity Assays (51chromium release assay)
Cell cytotoxicity was performed by standard 4-hour 51Cr release assay as previously described.Citation10 Effector peripheral blood mononuclear cells (PBMC) were isolated from buffy coats purchased from the New York Blood Center. PBMCs were cultured in vitro in medium either without (Medium) or with soluble IL15 or different IL15/IL15Rα complexes. After 72 hr of culture, the PBMCs were harvested, re-adjusted in numbers and tested for cytotoxicity against M14 human melanoma cells either in the absence or presence of different antibodies.
In vivo therapy studies
All animal procedures were performed in compliance with Institutional Animal Care and Use Committee (IACUC) guidelines. For in vivo therapy studies, BALB-Rag2-/-IL2R-γc-KO (DKO) mice (from Taconic as CIEA BRG mice)Citation10 or C57BL/6 mice (The Jackson Laboratory) were used. Effector PBMCs were prepared as described above. Prior to every experimental procedure, PBMCs were analyzed by flow cytometry for relative percentage of CD3, CD4, CD8 and CD56 cells to ensure consistency. Subcutaneous (sc) xenografts were created by implanting the tumor cells suspended in Matrigel (Corning Corp) in the flank of mice. Treatments were started when sc tumor size was reached 50–100 mm3 in general, or once successful iv tumor engraftment was confirmed by bioluminescence. Treatment schedules were marked on the figures, and doses of IL15 complexes, antibodies and effector cells were detailed in the Results section. Tumor size was measured using 1) hand-held TM900 scanner (Pieira, Brussels, BE), 2) Calipers, or 3) Bioluminescence. Bioluminescence imaging was conducted using the Xenogen In Vivo Imaging System (IVIS) 200 (Caliper LifeSciences). Briefly, mice were injected intravenously (iv) with 0.1 mL solution of D-luciferin (Gold Biotechnology; 30 mg/mL stock in PBS). Images were collected 1 to 2 minutes after injection using the following parameters: a 10- to 60-second exposure time, medium binning, and an 8 f/stop. Bioluminescence image analysis was performed using Living Image 2.6 (Caliper LifeSciences).
In vivo lymphocyte expansion studies
The lymphocyte expansion studies were conducted in healthy C57BL/6 mice injected sc one time with 5 µg WT-FL, or equimolar amount of MUT-SU (as positive control). Peripheral blood was collected on day 5 after the dosing, and CBC and FACS analysis were performed. Absolute cell count was calculated by multiplying the white blood cell count from CBC and percentage of positive cells from FACS.
Pharmacokinetics studies
Blood was collected by retro-orbital bleeding of DKO mice at time 0.5, 1, 2, 4, 8, 12, 24, 48 and 72 hr after a single sc injection of 50 ug of WT-FL or equimolar amount of other complexes. Concentrations of IL15 complexes in mouse sera were quantified by ELISA as described in the Cloning section. Pharmacokinetic analysis was carried out by non-compartmental analysis of the serum concentration-time data using Phoenix WinNonlin software program (Certara, Princeton, NJ).
Statistical analysis
Differences between samples were tested for significance by two-way ANOVA using Prism 7.0. p< .05 was considered statistically significant and indicated with * in the figures; p > .05 was considered not statistically significant and indicated with ns in some figures as well.
Results
Construction of IL15/IL15Rα-Fc complex
In order to most effectively compare the role of full-length IL15Rα (compared to the truncated sushi domain) or affinity matured IL15 (N72D, compared to the wildtype sequence), we prepared four different IL15/IL15Rα Fc-fusion proteins (): WT-FL which kept the wildtype IL15 sequence and full-length IL15Rα; MUT-FL which combined the N72D mutated IL15 with a full-length IL15Rα; WT-SU which combined the wildtype IL15 with the truncated sushi domain of IL15Rα; and MUT-SU which combined N72D IL15 and IL15Rα sushi domains (exact same amino acid coding sequence as ALT-803). All four proteins were fused the IL15Rα domain directly to the CH2-CH3 of human IgG1 Fc domain. After co-expression in CHO-S cells, all four complexes (WT-FL, MUT-FL, WT-SU and MUT-SU) were purified using Protein A affinity chromatography, with high protein yield (0.3–1.2 g/L).
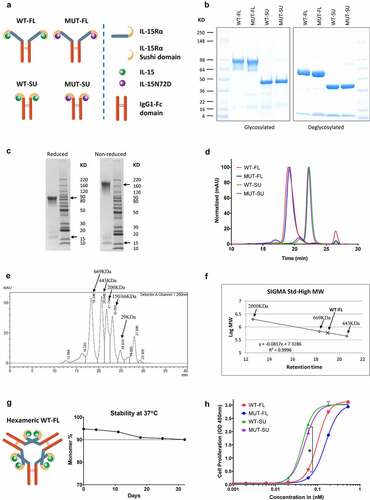
Figure 1. In vitro characterization of IL15/IL15Rα-Fc complexes. (a) Schematic diagrams of all four IL15/IL15Rα-Fc complexes in theoretical dimeric Fc fusion formats. (b) All four complexes were shown on reduced SDS-PAGE, before and after deglycosylation treatment, using Invitrogen SeeBlue Plus2 Pre-Stained Standard as the protein molecular weight (MW) marker. (c) WT-FL complex on SDS-PAGE under reduced and non-reduced conditions, using Life Technologies BenchMark Unstained Protein Ladder as the protein MW marker (the size of selected bands was shown), with IL15Rα-Fc and IL15 bands were marked with arrows. (d) SE-HPLC chromatography using G4000SW column. Major peak of WT-FL (19.0 minutes) corresponding to MW around 600 kDa, and SU complexes (22.3 minutes) corresponding to around 120 kDa, and salt buffer peak (26.5 minutes). (e) SE-HPLC results of the 6 standard MW marker (Sigma Gel Filtration Markers Kit) mixed together. Each MW marker was also run separately to confirm the position, except for the 150 and 66 kDa markers which did not separate in the mixture. (f) The fitted curve for WT-FL complex MW calculation. The size of 600 kDa of the WT-FL complex was calculated based on the fitted curve derived from the top three high MW standard markers that was run separately. (g) Schematic diagram of WT-FL complex in hexameric format, and accelerated stability test of WT-FL at 37°C over 4 weeks; monomer % represents the percentage of monomers in HPLC profile for each time point, based on AUC analysis excluding buffer peak. (h) In vitro cell proliferation assays using mouse lymphoblast cell line CTLL-2. Mean + SD (n = 3). * p < .0001 when FL-complexes treatment groups were compared with SU-complexes treatment groups at indicated concentration, respectively
In vitro biochemical characterization of IL15/IL15Rα-Fc complex
Under reducing conditions, WT-FL and MUT-FL complex gave rise to two major bands on Tris-glycine SDS-PAGE: ~60-90 KDa (IL15Rα-Fc) and ~16 KDa (IL15) (). The diffuse band pattern of IL15Rα-Fc was secondary to the heterogeneity of glycosylation of the full-length IL15Rα ectodomain in mammalian cells,Citation23 since it collapsed to the correct size of a single band at ~60 KDa after removal of sugar side chains by deglycosylation (). Similarly, the broad IL15 band also became more homogeneous (~13 kDa) with deglycosylation. In contrast, WT-SU and MUT-SU complexes migrated as two distinct bands, unaffected by deglycosylation because of their lower carbohydrate content (). To estimate the molecular size of the WT-FL complex more precisely, a BenchMark Unstained Protein Ladder was used for SDS-PAGE. Under reducing conditions, the WT-FL complex separated into monomers of around 100 KDa total (85 KDa for IL15Rα-Fc + 16 KDa for IL15, ); under non-reducing conditions where the disulfide bonds between Fc remained intact, the WT-FL complex separated into dimers of ~200 KDa total (170 KDa for dimeric IL15Rα-Fc + 2 × 16 KDa for two monomeric IL15, ).
SEC-HPLC analysis of purified WT-FL complexes showed a major peak at 600 KDa, with some minor aggregates removable by gel filtration (). The peak also appeared wide, consistent with the heterogeneous glycosylation. The size of 600 KDa of the WT-FL complex (19.0 minutes) was calculated based on high MW standard markers (), including a separately run 2000 KDa marker (run separate to reduce interference with the migration of other standards) (). Given the 200 kDa size of WT-FL dimer on SDS-PAGE, the 600 KDa size suggested that each dimeric IL15/IL15Rα-Fc dimer was trimerizing, forming a hexameric WT-FL complex (). In contrast, WT-SU and MUT-SU complexes formed stable dimeric Fc fusion complexes as expected (~120 KDa, ), implicating regions in the full length IL15Rα other than sushi domain are responsible for formation of the stable hexamer. This trimeric formation was also supported by the known crystal structure of IL15/IL15Rα complex (aa1-102, including sushi domain and adjacent linker region) forming a rod-like dimer, where the contact was between the two IL15Rα molecules.Citation24 Protein purity of all four complexes was determined to be over 90% by SEC-HPLC (WT-FL at 94%, MUT-FL at 95%, WT-SU at 92%, and MUT-SU at 90%). These complexes remained stable by SDS-PAGE and HPLC after multiple freeze and thaw cycles, and extended incubations at 37°C (HPLC data for WT-FL shown in ).
Effects of IL15/IL15Rα-Fc complexes on in vitro cell proliferation
The biological activity of these complexes was evaluated with cell proliferation assays using mouse lymphoblast cell line CTLL-2, which is known to respond to human IL15.Citation20 After incubating with IL15/IL15Rα-Fc complex for 3 days, CTLL-2 cell proliferation was measured using WST-8 kit. Although all four complexes were able to promote the growth of CTLL-2 cells in a dose-dependent manner, WT-SU and MUT-SU showed superior potency over WT-FL and MUT-FL ().
Binding of hu3F8 and hu3F8-BsAb to IL15/IL15Rα-stimulated lymphocyte subsets
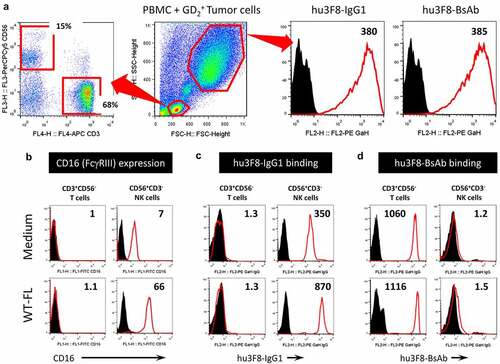
To address whether IL15/IL15Rα complexes can potentiate anti-tumor effects of hu3F8 antibodies, first we need to test if in vitro stimulation of human PBMCs with IL15/IL15Rα complexes could enhance binding of these antibodies to NK cells or T cells in the presence of tumor cells. When PBMC from healthy donors were incubated in the presence of GD2(+) M14 tumor cells, CD3+CD56− T cells and CD3−CD56+ NK cells could be clearly defined by FACS analysis (, left panel); here hu3F8-IgG1 (wild type Fc) and hu3F8-BsAb (Fc-silenced by N297A + K322A) bound equally well to tumor cells (, right panel). CD16 expression on NK cells was increased after culture at 37°C for 72 hrs in the presence of WT-FL complex (), resulting in increased binding of hu3F8-IgG1 via FcγRIII to CD16-expressng NK cells (). On the other hand, binding of hu3F8-BsAb via CD3 to CD3-expressing T cells remained high irrespective of whether PBMCs were cultured with WT-FL complex or not (). In summary, hu3F8-IgG1 could engage Fc-expressing NK cells which increased in the presence of WT-FL complex, while hu3F8-BsAb could engage CD3-expressing T cells.
Figure 2. In vitro binding of hu3F8 and hu3F8-BsAb to IL15-stimulated lymphocyte subsets. (a) Representative immunophenotypic appearance of normal PBMC-derived CD3+CD56− T cells and CD3−CD56+ NK cells (left panel). Representative flow cytometric appearance of PBMC and M14 tumor cells mixed together at 1:1 ratio (middle panel). Binding of hu3F8 and hu3F8-BsAb to M14 tumor cells (right two panels). (b) Expression of CD16 (FcγRIII) on T cells and NK cells. (c) Binding of hu3F8 IgG1 via FcγRIII to CD16-expressing NK cells but not T cells. (d) Binding of hu3F8-BsAb via CD3 to CD3-expressing T cells but not NK cells. PBMCs were cultured in complete RPMI medium either without (Medium) or with 1 nM WT-FL complex for 72 hrs before FACS analysis. Numbers in right upper corner of each histogram box represent MFI ratios calculated as geo-MFI of antigen-specific staining (red-line histogram) divided by geo-MFI of isotype-control IgG staining (black filled peak)
Stimulation of PBMC with IL15/IL15Rα-Fc complex augmented in vitro tumor cytotoxicity
To test if in vitro stimulation of human PBMCs with soluble IL15 or IL15/IL15Rα complexes could augment anti-tumor cytotoxicity, fresh PBMCs were primed for 72 hrs in increasing concentrations of soluble IL15, or IL15/IL15Rα complexes, followed by a 4-hour in vitro cytotoxicity assay against GD2(+) human melanoma cell line M14. As shown in , at highest concentration (1 nM), all five formats (free IL15 or four different IL15/IL15Rα complexes) were equally potent in activating lymphocytes to mediate M14 tumor lysis; however, at lower concentrations, there was a significant enhancement in lymphocyte activation by MUT-SU and MUT-FL complexes versus WT-SU and WT-FL complexes. Such results were anticipated since improved binding of mutant IL15 to IL2Rβγc should result in stronger signaling, making PBMC derived T- and NK cells more cytolytic. The absence of significant difference between soluble IL15 versus WT-FL or WT-SU complexes could be explained by the presence of native IL15Rα on accessory cells (non-T, non-NK) within the PBMC population that presented IL15 to T- and NK cells.
Figure 3. In vitro cytotoxicity by IL15-stimulated PBMCs. PBMCs from one healthy donor was cultured in vitro in medium either without (Medium) or with soluble IL15 or different IL15/IL15Rα complexes. After 72 hrs of culture, the PBMCs were harvested, re-adjusted in numbers and tested in an in vitro cytotoxicity (51Cr-release) assay against M14 human melanoma cells either in the absence or presence of different antibodies. Results are presented as percentage of tumor cell lysis (Mean + SEM, n = 3). * p < .01 when WT-complexes treatment groups were compared with MUT-complexes treatment groups at indicated concentration or E:T ratio, respectively. (a) Antibody-independent cytotoxicity titrated by IL15 concentrations. E:T ratio at 10:1. (b) Antibody-dependent cytotoxicity titrated by hu3F8 (middle panel) or hu3F8-BsAb (right panel). IL15 complexes concentration at 0.001 nM, and E:T ratio at 10:1. (c) Cytotoxicity titrated by E:T ratios. IL15 complexes concentration at 1 nM, and antibodies concentration at 0.01 ug/mL
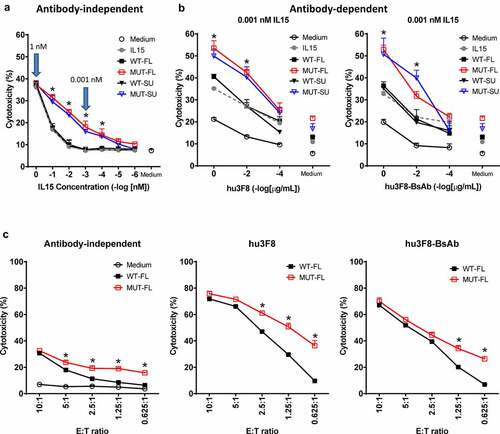
Next, we tested if IL15-primed PBMC could potentiate antibody-dependent cell-mediated cytotoxicity (ADCC) in the presence of hu3F8-IgG1 (naxitamab) or antibody-dependent T cell-mediated cytotoxicity (ADTC) in the presence hu3F8-BsAb. As shown in , cytotoxicity by IL15-stimulated PBMC () was substantially improved in the presence of anti-tumor antibodies, an effect that was antibody concentration-dependent, even at low concentrations (0.001 nM) of the complex. Consistent with the antibody-independent dataset, incubation with MUT-FL or MUT-SU complexes resulted in greater cytotoxicity compared to WT-FL or WT-SU complexes or soluble IL15. Furthermore, since there was no substantial difference between FL and SU platforms (either MUT-FL vs MUT-SU or WT-FL vs WT-SU), we focused our comparison between WT-FL and MUT-FL complexes. To see how priming with IL15 impacted cytotoxicity at low numbers of effectors, E:T ratios was titrated down while keeping the concentrations constant for IL15 complexes at 1 nM and antibodies at 0.01 ug/mL. Consistent with the previous data, MUT-FL stimulated cytotoxicity in PBMC better than WT-FL (), both in the absence of antibodies, and in the presence of hu3F8, or hu3F8-BsAb. Similar patterns of activation were seen with five different PBMC donors (Supplemental Figure S1). In summary, MUT-FL and MUT-SU complexes provided better stimulation to naïve PMBCs and augmented tumor cytotoxicity than WT-FL or WT-SU complexes in vitro.
Immunophenotypic changes of PBMC induced by WT-FL versus MUT-FL complexes
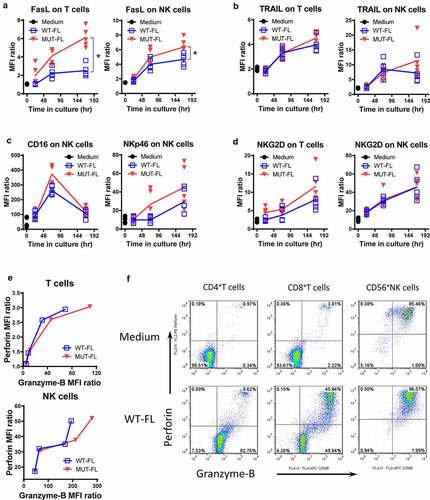
To assess the changes in phenotype or function of T cells and NK cells following incubation with WT-FL or MUT-FL, PBMC were cultured in the presence of IL15/IL15Rα complexes between 24 and 168 hr. Both WT-FL and MUT-FL stimulated time-dependent upregulation of death ligands FasL and TRAIL, on T and NK cells, however, a difference between WT-FL vs. MUT-FL complexes was observed only for FasL but not for TRAIL (). Activating receptors NKp46 and NKG2D both displayed upregulation in response to incubation with WT-FL or MUT-FL, however, no significant difference was seen between complexes. Consistent with previous reports, CD16 (FcγRIII) expression also increased with incubation, peaking at 72 hr followed by a drop by 168 hrCitation25 (), a pattern consistently observed with PBMCs from five different donors. Interestingly, whereas no changes in KIR2DL1, KIR3DL1 and NKG2A, the inhibitory receptors on NK cells, were observed even after extended (up to 168 hr) exposure to IL15/IL15Rα complexes (Supplemental Figure S2A), the inhibitory/exhaustion markers on CD3+ T cells (PD1, TIM3 and LAG3) were strongly upregulated (Supplemental Figure S2B). This provides a rationale for future combination study of IL15 complex with immune checkpoint blockade. Besides, whereas WT-FL and MUT-FL induced strong proliferation for both CD8+ T cells and CD4+FoxP3- T cells over 168 hr period, almost no proliferation was observed for CD4+FoxP3+ Tregs cells, especially for WT-FL complex (Supplemental Figure S2C), consistent with the published data from the ALT-803 clinical trials.Citation25 While naïve T cells displayed substantial upregulation in granzyme-B following stimulation with WT-FL or MUT-FL, naïve NK cells markedly upregulated both perforin and granzyme-B above their already high baseline levels (). These results suggest that following stimulation via IL2Rβγc both T- and NK cells become activated as measured by upregulated expression of stimulatory receptors, cytotoxic proteins both intra-cytoplasmically and on the cell surface. In general, MUT-FL provides better stimulation to naïve PMBCs than WT-FL, which is consistent with the in vitro cytotoxicity.
Figure 4. In vitro immunophenotype of IL15-stimulated PBMCs. PBMCs from healthy donors (n = 5) were cultured in vitro in medium either without (Medium) or with 1 nM WT-FL or MUT-FL complex. At 24, 72 and 168 hr cells were tested by flow cytometry for expression of different surface markers. Analysis is based on gating on T cells (CD3+CD56− lymphocytes) or NK cells (CD3−CD56+ lymphocytes). Results are presented as geo-MFI ratio of the marker of interest, individual for each donor. Lines represent Mean (n = 5) of those 5 individual values. * p < .01 when WT-FL treatment groups were compared with MUT-FL treatment groups at indicated time point, respectively. (a) Surface expression of FasL. (b) Surface expression of TRAIL. (c) Surface expression of CD16 and NKp46. (d) Surface expression of NKG2D. (e) Intracellular co-expression of Perforin and Granzyme-B. PBMCs from one healthy donor was used in this case. Results are presented as geo-MFI ratio of the marker of interest, at each time point of testing. To enable Perforin and Granzyme-B retention, Brefeldin-A was added to cultures for the last 6 hr of each time point. Representative FACS dot-plots at 72 hr time point were shown in (f)
WT-FL IL15 complex promotes in vivo lymphocyte expansion and improves serum half-life in mice
To determine the effect of WT-FL IL15 complex on lymphocyte expansion in vivo, we administered the complex one time subcutaneously in tumor-free C57BL/6 mice injected with 5 ug WT-FL, or 4 ug MUT-SU (equimolar amount, as positive control).Citation26 Peripheral blood was collected on day 5 after the dosing, and CBC and FACS analysis were performed. Absolute cell counts were calculated by multiplying the white blood cell count from CBC and the percentage of positive cells from FACS. As shown in , WT-FL complex promoted both CD3+ T cell (mainly CD8+ T cells) and NK1.1+ NK cell expansion. Whereas WT-FL promoted a bit more NK cell expansion, T cell expansion was significantly greater with MUT-SU, which was consistent with the in vitro CTLL-2 cell proliferation results ().
Figure 5. WT-FL IL15 complex promotes in vivo lymphocyte expansion and improves serum half-life in mice. (a) Lymphocyte expansion in healthy C57BL/6 mice. Absolute cell counts were calculated by multiplying the white blood cell count from CBC and percentage of positive cells from FACS. Mean ± SEM (n = 5). * p < .001 when WT-FL treatment group was compared with either no treatment (PBS) group or MUT-SU treatment group, respectively; ns, p > .05 when WT-FL treatment group was compared with MUT-SU treatment group. (b) Pharmacokinetics of IL15 complexes. Quantitation of serum IL15 complexes was carried out by ELISA, and the data were depicted using GraphPad Prism software. Mean + SD (n = 5). Pharmacokinetic analysis was carried out by non-compartmental analysis of the serum concentration-time data using WinNonlin software program, and presented in the inserted Table
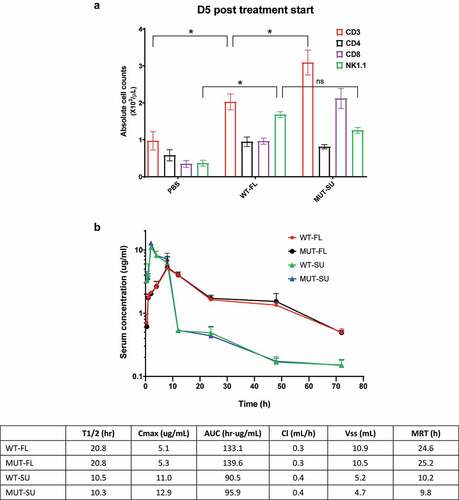
Pharmacokinetics studies of all four complexes in mice were also performed. 50 ug of FL-complexes or 40 ug SU-complexes were injected subcutaneously into DKO mice, and concentration of IL15 complexes in mouse sera were quantified by ELISA. Pharmacokinetics analysis was carried out by the non-compartmental model. As shown in when compared to SU-based complexes, FL-based complexes had lower peak serum levels (Cmax), longer serum half-life (T1/2), greater drug exposure (AUC), and longer mean residence time (MRT). Since the in vivo effects of IL15 is not immediate following subcutaneous administration, the effect of a delayed drug exposure of IL15 complex beyond peak time (8 hrs) may be further amplified, based on AUC differences after peak time (8–72 hrs) between FL-complexes (~110 hr·ug/mL) and SU-complexes (~30 hr·ug/mL).
WT-FL IL15 complex potentiates anti-tumor effects of antibody immunotherapy against tumor cell line xenografts
To establish how WT-FL impacted in vivo anti-tumor function, we tested it in four separate tumor models: (i) an intravenous neuroblastoma xenograft model using immunodeficient mice (BALB-Rag2-/-IL2R-γc-KO, DKO) treated with BsAb, (ii) a subcutaneous neuroblastoma xenograft model using DKO mice treated with IgG, (iii) a subcutaneous melanoma xenograft model using DKO mice treated with BsAb, and (iv) a C57BL/6 immuno-competent mouse model bearing a subcutaneous GD2(+) melanoma treated with IgG.
To begin, we tested the impact of WT-FL on BsAb-mediated anti-tumor function using mice bearing intravenous tumors. 0.5 × 106 GD2(+) IMR32 neuroblastoma cells were infused intravenously to mice to model metastatic disease, which primarily resided and propagated in liver and lymph nodes, and treatment began once successful tumor engraftment was confirmed by bioluminescence. Hu3F8-BsAb has been shown to be effective at 40 ug dose previously.Citation10 In order to see the potentiation effect of IL15 complexes, mice were treated with subtherapeutic doses of hu3F8-BsAb starting on day 12 (10 µg iv, 2x/wk x 3 wks), PBMCs were administered starting on day 14 (5x106 iv, q wk x 2 wks) and IL15 complexes were administered sc on day 14 (10 ug of FL complexes or 8 ug of SU complexes, 2x/wk x 3 wks). Tumor bioluminescence was measured weekly. In contrast to the in vitro data, only WT-FL and MUT-FL treated mice were able to eradicate tumor growth, while mice treated with hu3F8-BsAb alone, or in combination with WT-SU or MUT-SU had a marginal impact on tumor growth ().
Figure 6. WT-FL IL15 complex potentiates anti-tumor effects of antibody immunotherapy against tumor cell line xenografts. Treatment schedules were marked on the figures, and doses of IL15 complexes, antibodies and effector cells were detailed in the Results. Data shown as mean + SEM (n = 5); (a-b) iv tumor plus iv effector cells model: Bioluminescence changes of IMR32 neuroblastoma during treatment (a) and representative images at day 25 (b). * p < .05 when FL-complexes treatment groups were compared with all other four groups at indicated time point, respectively. (c) sc tumor plus sc effector cells (1:1 mixing) model: tumor volume changes of IMR32 neuroblastoma. * p < .05 when FL-complexes treatment groups were compared with No treatment group at indicated time point, respectively; ns, p > .05 when SU-complexes treatment groups were compared with No treatment group at indicated time point, respectively. (d) sc tumor plus iv effector cells model: % tumor growth (calculated as tumor volume at indicated time point divided by tumor volume at Day 8 before the treatment start) of M14 melanoma. * p < .0001 when FL-complexes treatment groups were compared with all other four groups at indicated time point, respectively. (e) C57BL/6 mice grafted with sc GD2(+) murine melanoma cells: tumor volume changes of B78/D14 melanoma. * p < .0001 when FL-complexes treatment groups were compared with SU-complexes treatment groups at indicated time point, respectively
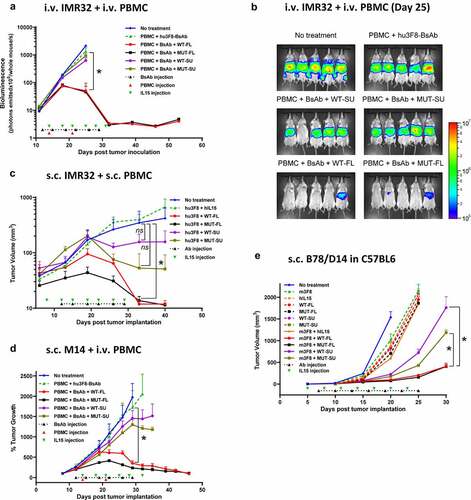
To test the role of IL15 complexes on solid tumors bearing infiltrated lymphocytes, we tested all formats in DKO mice bearing subcutaneous IMR32 neuroblastoma tumors mixed with PBMCs (1:1, 5 × 106 cells each). Mice were treated with hu3F8-IgG1 starting on day 12 (50 ug iv, 2x/wk x 3 wks), plus iv IL15 complexes starting on day 8 (10 ug of FL complexes, 8 µg of SU complexes or 2.25 ug of hIL15, 2x/wk for total 7 doses). Both WT-FL and MUT-FL complex in combination with hu3F8-IgG1 were able to suppress tumor growth, and to a greater extent than either recombinant hIL15, WT-SU or MUT-SU complexes ().
Next, we assessed the role of IL15 complexes on the treatment of solid tumors where lymphocytes had to home to the tumor. Here 4 × 106 M14 melanoma cells were implanted subcutaneously into DKO mice. Once tumors were engrafted, mice began treatment with low doses of hu3F8-BsAb (10–20 ug iv, 2x/wk x 3 wks), PBMCs (5x106 iv, q wk x 2 wks) and sc IL15 complexes (10 ug of FL complexes or 8 ug of SU complexes, 2x/wk x 3 wks). Tumor size was measured by calipers twice per week. Consistent with the intravenous tumor model, only WT-FL and MUT-FL treated mice eradicated tumors, while hu3F8-BsAb alone or in combination with WT-SU or MUT-SU only slowed tumor growth ().
To evaluate the effect of each IL15 complex in an immunocompetent model, C57BL/6 mice bearing 1 × 105 GD2(+) B78/D14 melanoma tumor cells implanted subcutaneously were treated with anti-GD2 m3F8-IgG3 and sc IL15 complexes, starting on day 7 (50 ug iv, 2x/wk for total 7 doses), and day 5 (2 ug of FL complexes, 1.6 µg of SU complexes or 0.45 ug of hIL15, 2x/wk for total 7 doses), respectively. In the presence of m3F8, both WT-FL and MUT-FL were able to strongly suppress tumor growth, while WT-SU and MUT-SU only showed a modest anti-tumor effect. Importantly, neither m3F8 nor any of the IL15 complexes alone had any significant impact on tumor growth, suggesting WT-FL and MUT-FL were both synergizing with m3F8 ().
In summary, WT-FL and MUT-FL constructs consistently showed superior in vivo potency than the SU-based WT-SU and MUT-SU complexes. Although this was not predicted from the in vitro incubation data, it did correlate with the improved pharmacokinetic profile (see discussion below), suggesting that the unique multimerization of the WT-FL and MUT-FL formats has a role in improving activity.
Discussion
In this study, we described the successful engineering of a full-length Fc-dimerized IL15/IL15Rα complex (WT-FL) that formed spontaneous trimers of dimers (6 IL15 + 6 IL15Rα). The complex was able to stimulate PBMC-derived T cells and NK cells and increase their in vitro tumor cytotoxicity, and elicit expression of surrogate markers of activation and cytotoxicity. Moreover, using MUT-SU (ALT-803) as our benchmark, the hexameric WT-FL proved to be substantially more potent in vivo. In xenograft models, WT-FL demonstrated longer serum half-life, and enhanced anti-neuroblastoma and anti-melanoma cytotoxicity in the presence of NK cells + IgG antibodies, or T cells + BsAb.
In preclinical studies, ALT-803 was synergistic in ADCC with other therapeutic antibodies and immune checkpoint inhibitors, such as anti-CD20, anti-PD1, anti-PDL1 and anti-CTLA4 antibodies.Citation26,Citation27 ALT-803 could even be used as a scaffold when fused to single chains of the tumor-targeting monoclonal antibody rituximab, and demonstrated antigen-specific anti-tumor responses.Citation28 ALT-803 has been proven safe and effective in human clinical trials (NCT01885897).Citation25 Other phase I/II trials of ALT-803 are ongoing for solid tumors, hematological malignancies and HIV, either as monotherapy or in combination with other therapeutic antibodies, immune checkpoint inhibitors, or chemotherapy drugs. Most recently, ALT-803 (renamed as N-803) when combined with BCG showed promising responses in BCG-unresponsive non-muscle invasive bladder cancer, convincing the FDA to grant Breakthrough Therapy Designation in 2019.Citation29 Here, we showed that WT-FL complex synergized with not only anti-tumor IgG antibodies but also T cell engaging bispecific antibodies, implicating potentially broad clinical applications. The complex could be used in combination with NK cells-engaging antibodies or T cell-engaging bispecific antibodies in cancer immunotherapy, and with probably other NK or T cell-based therapeutics, such as chimeric antigen receptor-modified T cells and immune checkpoint inhibitor. A formal GLP toxicology study of WT-FL IL15 complex in mice completed recently confirmed the lack of biochemical and pathological toxicities of these constructs at clinically relevant dosages. Human toxicities will have to await phase I clinical trial.
The spontaneous multimerization of the WT-FL fusion protein to a hexamer was unexpected, but likely important property driving its enhanced activity in vivo. This is distinct from the MUT-SU format (ALT-803), which only dimerizes (2 IL15 + 2 IL15Rα), or the full-length heterodimeric IL15/IL15Rα complex (hetIL15),Citation30 which has no Fc domain and only forms a monomer (1 IL15 + 1 IL15Rα). How this hexamer of WT-FL forms is not yet clear, but it seems that both Fc-Fc interaction and IL15Rα-IL15Rα interaction beyond sushi domains are required. This hexameric complex may even provide enhanced stability over other multimers, analogous to the hexameric formation of IgG-Complement complex on the cell surface.Citation31 More importantly with a prolonged half-life, the WT-FL could also be administered less frequently which could be conducive to better compliance.
Previous studies have suggested that the soluble full-length ectodomain IL15Rα was antagonistic while the Sushi domain was not.Citation32 Another important and unexpected finding from the current study relates to the role of the N72D mutation in IL15. Our data showed that complexes carrying the mutated form of IL15 (MUT-SU and MUT-FL) had stronger in vitro potency over complexes carrying the wild-type IL15 (WT-SU and WT-FL) ( and ), yet did not translate into superiority in vivo, where the WT-FL and MUT-FL consistently demonstrated the most potent activity (). This could partly be explained by the differential pharmacokinetics in mice, where the FL-based complexes displayed longer serum half-life, higher AUC, and longer mean residence time compared to the SU-based complexes (). Additionally, while MUT-SU complex was indeed better than WT-SU as previously reported, the difference between MUT-FL and WT-FL was less obvious. This discordance between in vitro potency and in vivo efficacy suggest a more complex biology that likely involve the differential effects of IL15 constructs on effector cell homing and cooperation with other cell types. In fact, the recent report using potency-reduced IL15 complex (XmAb24306 from Xencor) to improve in vivo efficacy over the WT counterpartCitation33 supported the advantage of a prolonged half-life. If we used in vitro proliferation as a criteria of in vitro potency as was done with XmAb24306, our FL-based complexes could be considered as potency-reduced (). The finding that WT-FL and MUT-FL performed similarly in vivo (no statistically significant difference) was also unexpected, given the previous data suggesting IL15 N72D enhanced cell binding. Since mutations can create new epitopes for both T cells and B cells thereby risking immunogenicity of a therapeutics, their avoidance in WT-FL has at least a theoretical advantage. Plus, MUT-FL induced mild Tregs proliferation whereas WT-FL did not (p < .01, Supplemental Figure S2C), which gives WT-FL another important advantage over MUT-FL for future clinical development.
In this manuscript, we chose GD2 and neuroblastoma as a tumor model.Citation5 GD2 is an adhesion molecule abundantly expressed on neuroblastoma. It is also widely expressed on melanomas, small cell lung cancers, bone or soft tissue sarcomas, retinoblastomas and certain brain tumors. At least three anti-GD2 monoclonal antibodies underwent extensive clinical testing, i.e. murine 3F8 (m3F8),Citation34 chimeric 14.18 (ch14.18, dinutuximab)Citation35 and humanized 3F8 (naxitamab).Citation36,Citation37 In a phase III randomized clinical trial led by the Children’s Oncology Group, dinutuximab in combination with interleukin-2 (IL2) and GM-CSF produced a statistically significant improvement in overall survival (OS).Citation38,Citation39 Chimeric antigen receptor-modified effectors, e.g. anti-GD2 CARTCitation5,Citation40 and CAR-NKT,Citation41 are active areas of clinical investigation where cell persistence in vivo has been major challenges. As a pro-survival growth factor, IL15 is well suited to sustain adoptive NK cell and T cell immunotherapy,Citation42 where poor effector cell persistence has been one of the major limitations. IL15/IL15Rα complex increases proliferation of both NK and CD8+/CD44high T cells in vitro and NK killing in vivo,Citation43 suppresses established murine solid tumors,Citation44 and induces specific anti-tumor immunity when injected intratumorallyCitation45 and in neuroblastoma, IL15 can rescue cells from M2 macrophage-induced cell death.Citation46 Recent work using a premixed murine IL15/IL15Rα complex was also shown to enhance anti-GD2 antibody therapy in patient-derived neuroblastoma xenografts.Citation47
In summary, we demonstrated that a WT-FL IL15/IL15Rα-Fc fusion complex increased serum half-life and enhanced the anti-tumor effect of FcR-dependent and T cell-based immunotherapies in vivo. This enhancement appears related to its hexameric form, an unexpected consequence of using the fully wild-type IL15 and IL15Rα sequences. While previous studies suggested the soluble full-length ectodomain IL15Rα be inhibitory instead of stimulatory on immune cells,Citation32 our findings proved that not only was WT-FL stimulatory, but it showed enhanced anti-tumor activity over the MUT-SU format. These results support continued study of this complex for clinical development.
Disclosure
HX and NKC were named as co-inventors in patents on IL15/IL15Rα-Fc complex, and hu3F8 antibodies filed by MSK. NKC reports receiving commercial research grants from Y-mAbs Therapeutics and Abpro-Labs Inc.; holding ownership interest/equity in Y-mAbs Therapeutics Inc., holding ownership interest/equity in Abpro-Labs, and owning stock options in Eureka Therapeutics. NKC is the inventor and owner of issued patents licensed by MSK to Y-mAbs Therapeutics, Biotec Pharmacon, and Abpro-labs. Hu3F8 and 8H9 were licensed by MSKCC to Y-mAbs therapeutics. Both MSK and NKC have financial interest in Y-mAbs. NKC is an advisory board member for Abpro-Labs and Eureka Therapeutics. The other authors declare they have no competing interests as defined by the Journal, or other interests that might be perceived to influence the results and discussion reported in this paper.
Supplemental Material
Download ()Acknowledgments
We thank MSK Small-Animal Imaging Core Facility, and Laboratory of Comparative Pathology for providing technical services, Dr Irene Cheung and Dr Brian Santich for reviewing the manuscript.
Funding
This study was supported in part by grants from the following: Lebara Foundation, Kids Walk for Kids with Cancer NYC, Robert Steel Foundation, and Enid A. Haupt Chair Endowment Fund. Technical service provided by the MSK Animal Imaging Core Facility was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–14. doi:10.1038/nature10673.
- Raffaghello L, Prigione I, Airoldi I, Camoriano M, Morandi F, Bocca P, Gambini C, Ferrone S, Pistoia V. Mechanisms of immune evasion of human neuroblastoma. Cancer Lett. 2005;228(1–2):155–161. doi:10.1016/j.canlet.2004.11.064
- Coughlin CM, Fleming MD, Carroll RG, Pawel BR, Hogarty MD, Shan X, Vance BA, Cohen JN, Jairaj S, Lord EM, et al. Immunosurveillance and survivin-specific T-cell immunity in children with high-risk neuroblastoma. J Clin Oncol. 2006;24:5725–5734
- Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer. 2013;13(6):397–411. doi:10.1038/nrc3526.
- Park JA, Cheung NK. Targets and Antibody Formats for Immunotherapy of Neuroblastoma. Journal of Clinical Oncology. 2020; 19:01410. doi:10.1200/jco.19.01410
- Park JA, Cheung NV. Limitations and opportunities for immune checkpoint inhibitors in pediatric malignancies. Cancer Treat Rev. 2017;58:22–33. doi:10.1016/j.ctrv.2017.05.006.
- Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–1355. doi:10.1126/science.aar4060.
- Dondero A, Pastorino F, Della Chiesa M, Corrias MV, Morandi F, Pistoia V, Olive D, Bellora F, Locatelli F, Castellano A, et al. PD-L1 expression in metastatic neuroblastoma as an additional mechanism for limiting immune surveillance. OncoImmunology. 2016;5(1):e1064578. doi:10.1080/2162402X.2015.1064578
- Galluzzi L, Vacchelli E, Eggermont A, Fridman WH, Galon J, Sautes-Fridman C, Tartour E, Zitvogel L, Kroemer G. Trial Watch: adoptive cell transfer immunotherapy. OncoImmunology. 2012;1(3):306–315. doi:10.4161/onci.19549
- Xu H, Cheng M, Guo H, Chen Y, Huse M, Cheung NK. Retargeting T cells to GD2 pentasaccharide on human tumors using Bispecific humanized antibody. Cancer Immunology Research. 2015;3(3):266–277. doi:10.1158/2326-6066.CIR-14-0230-T.
- Waldmann TA, Miljkovic MD, Conlon KC. Interleukin-15 (dys)regulation of lymphoid homeostasis: implications for therapy of autoimmunity and cancer. J Exp Med. 2020;217(1):217. doi:10.1084/jem.20191062.
- Steel JC, Waldmann TA, Morris JC. Interleukin-15 biology and its therapeutic implications in cancer. Trends Pharmacol Sci. 2012;33(1):35–41. doi:10.1016/j.tips.2011.09.004.
- Berger C, Berger M, Hackman RC, Gough M, Elliott C, Jensen MC, Riddell SR. Safety and immunologic effects of IL-15 administration in nonhuman primates. Blood. 2009;114(12):2417–2426. doi:10.1182/blood-2008-12-189266
- Marks-Konczalik J, Dubois S, Losi JM, Sabzevari H, Yamada N, Feigenbaum L, Waldmann TA, Tagaya Y. IL-2-induced activation-induced cell death is inhibited in IL-15 transgenic mice. Proc Natl Acad Sci U S A. 2000;97(21):11445–11450. doi:10.1073/pnas.200363097
- Munger W, DeJoy SQ, Jeyaseelan R Sr., Torley LW, Grabstein KH, Eisenmann J, Paxton R, Cox T, Wick MM, Kerwar SS. Studies evaluating the antitumor activity and toxicity of interleukin-15, a new T cell growth factor: comparison with interleukin-2. Cell Immunol. 1995;165(2):289–293. doi:10.1006/cimm.1995.1216
- Dubois S, Mariner J, Waldmann TA, Tagaya YIL. 15Ralpha recycles and presents IL-15 In trans to neighboring cells. Immunity. 2002;17(5):537–547. doi:10.1016/S1074-7613(02)00429-6.
- Cheever MA. Twelve immunotherapy drugs that could cure cancers. Immunol Rev. 2008;222(1):357–368. doi:10.1111/j.1600-065X.2008.00604.x.
- Mohler KM, Torrance DS, Smith CA, Goodwin RG, Stremler KE, Fung VP, Madani H, Widmer MB. Soluble tumor necrosis factor (TNF) receptors are effective therapeutic agents in lethal endotoxemia and function simultaneously as both TNF carriers and TNF antagonists. J Immunol. 1993;151(3):1548–1561
- Han KP, Zhu X, Liu B, Jeng E, Kong L, Yovandich JL, Vyas VV, Marcus WD, Chavaillaz PA, Romero CA, et al. IL-15:IL-15 receptor alpha superagonist complex: high-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine. 2011;56(3):804–810. doi:10.1016/j.cyto.2011.09.028
- Zhu X, Marcus WD, Xu W, Lee HI, Han K, Egan JO, Yovandich JL, Rhode PR, Wong HC. Novel human interleukin-15 agonists. J Immunol. 2009;183(6):3598–3607. doi:10.4049/jimmunol.0901244
- Wu Z, Xu Y. IL-15R alpha-IgG1-Fc enhances IL-2 and IL-15 anti-tumor action through NK and CD8+ T cells proliferation and activation. J Mol Cell Biol. 2010;2:(4):217–222. doi:10.1093/jmcb/mjq012.
- Cheung NK, Modak S, Lin Y, Guo H, Zanzonico P, Chung J, Zuo Y, Sanderson J, Wilbert S, Theodore LJ, et al. Single-chain Fv-streptavidin substantially improved therapeutic index in multistep targeting directed at disialoganglioside GD2. J Nucl Med. 2004;45(5):867–877
- Thaysen-Andersen M, Chertova E, Bergamaschi C, Moh ES, Chertov O, Roser J, Sowder R, Bear J, Lifson J, Packer NH, et al. Recombinant human heterodimeric IL-15 complex displays extensive and reproducible N- and O-linked glycosylation. Glycoconj J. 2016;33(3):417–433. doi:10.1007/s10719-015-9627-1
- Chirifu M, Hayashi C, Nakamura T, Toma S, Shuto T, Kai H, Yamagata Y, Davis SJ, Ikemizu S. Crystal structure of the IL-15-IL-15Ralpha complex, a cytokine-receptor unit presented in trans. Nat Immunol. 2007;8: 1001–1007
- Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, Wagner JE, Weisdorf DJ, Blazar BR, Ustun C, DeFor TE, et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood. 2018;131: 2515–2527
- Rhode PR, Egan JO, Xu W, Hong H, Webb GM, Chen X, Liu B, Zhu X, Wen J, You L, et al. Comparison of the Superagonist Complex, ALT-803, to IL15 as Cancer Immunotherapeutics in Animal Models. Cancer Immunology Research. 2016;4(1):49–60. doi:10.1158/2326-6066.CIR-15-0093-T
- Rosario M, Liu B, Kong L, Collins LI, Schneider SE, Chen X, Han K, Jeng EK, Rhode PR, Leong JW, et al. The IL-15-Based ALT-803 Complex Enhances FcγRIIIa-Triggered NK Cell Responses and In Vivo Clearance of B Cell Lymphomas. Clin Cancer Res. 2016;22(3):596–608. doi:10.1158/1078-0432.CCR-15-1419
- Liu B, Kong L, Han K, Hong H, Marcus WD, Chen X, Jeng EK, Alter S, Zhu X, Rubinstein MP, et al. A Novel Fusion of ALT-803 (Interleukin (IL)-15 Superagonist) with an Antibody Demonstrates Antigen-specific Antitumor Responses. J Biol Chem. 2016;291:23869–23881
- Chamie K, Lee JH, Rock A, Rhode PR, Soon-Shiong P. Preliminary phase 2 clinical results of IL-15RαFc superagonist N-803 with BCG in BCG-unresponsive non-muscle invasive bladder cancer (NMIBC) patients. Journal of Clinical Oncology. 2019;37(15_suppl):4561. doi:10.1200/JCO.2019.37.15_suppl.4561.
- Chertova E, Bergamaschi C, Chertov O, Sowder R, Bear J, Roser JD, Beach RK, Lifson JD, Felber BK, Pavlakis GN. Characterization and favorable in vivo properties of heterodimeric soluble IL-15.IL-15Ralpha cytokine compared to IL-15 monomer. J Biol Chem. 2013;288(25):18093–18103. doi:10.1074/jbc.M113.461756
- Diebolder CA, Beurskens FJ, De Jong RN, Koning RI, Strumane K, Lindorfer MA, Voorhorst M, Ugurlar D, Rosati S, Heck AJ, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science. 2014;343(6176):1260–1263. doi:10.1126/science.1248943
- Mortier E, Quemener A, Vusio P, Lorenzen I, Boublik Y, Grotzinger J, Plet A, Jacques Y. Soluble interleukin-15 receptor alpha (IL-15R alpha)-sushi as a selective and potent agonist of IL-15 action through IL-15R beta/gamma. Hyperagonist IL-15 x IL-15R alpha fusion proteins. J Biol Chem. 2006;281(3):1612–1619. doi:10.1074/jbc.M508624200
- Bernett MJ, Varma R, Bonzon C, Rashid R, Bogaert L, Liu K, Schubbert S, Avery KN, Leung IWL, Rodriguez N, et al. Potency-reduced IL15/IL15Ra heterodimeric Fc-fusions display enhanced in vivo activity through increased exposure. AACR Annual Meeting 2018, Chicago, Illinois, USA. 2018; Poster # 5565
- Cheung NK, Saarinen UM, Neely JE, Landmeier B, Donovan D, Coccia PF. Monoclonal antibodies to a glycolipid antigen on human neuroblastoma cells. Cancer Res. 1985;45:2642–2649.
- Mujoo K, Kipps TJ, Yang HM, Cheresh DA, Wargalla U, Sander DJ, Reisfeld RA. Functional properties and effect on growth suppression of human neuroblastoma tumors by isotype switch variants of monoclonal antiganglioside GD2 antibody 14.18. Cancer Res. 1989;49: 2857–2861
- Kushner BH, Cheung IY, Modak S, Basu EM, Roberts SS, Cheung NK. Humanized 3F8 Anti-GD2 Monoclonal Antibody Dosing With Granulocyte-Macrophage Colony-Stimulating Factor in Patients With Resistant Neuroblastoma: A Phase 1 Clinical Trial. JAMA Oncol 20184:1729-1735
- Kushner BH, Modak M, Cheung IY, Basu EM, Kudva A, Roberts SS, Cheung NK. High-dose humanized-3F8 (hu3F8) plus stepped-up dosing of GM-CSF: outpatient treatment, low immunogenicity, and major responses in a phase II trial. Advances in Neuroblastoma Research Congress 2018. San Francisco, CA, USA. 2018; Abstract # 287
- Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363(14):1324–1334. doi:10.1056/NEJMoa0911123
- Yu AL, Gilman AL, Ozkaynak MF, Sondel PM, London WB. Update of outcome for high-risk neuroblastoma treated on a randomized trial of chimeric anti-GD2 antibody (ch14.18) + GM-CSF/IL2 immunotherapy in 1st response: a children’s oncology group study. Advances in Neuroblastoma Research Association 2014. Cologne (Germany) 2014
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270
- Heczey A, Courtney AN, Montalbano A, Robinson S, Liu K, Li M, Ghatwai N, Dakhova O, Liu B, Raveh-Sadka T, et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interim analysis. Nat Med. 1686–90;2020:26.
- Teague RM, Sather BD, Sacks JA, Huang MZ, Dossett ML, Morimoto J, Tan X, Sutton SE, Cooke MP, Ohlen C, et al. Interleukin-15 rescues tolerant CD8+ T cells for use in adoptive immunotherapy of establishedtumors. Nat Med. 2006;12(3):335–341. doi:10.1038/nm1359
- Dubois S, Patel HJ, Zhang M, Waldmann TA, Muller JR. Preassociation of IL-15 with IL-15Rα-IgG1-Fc Enhances Its Activity on Proliferation of NK and CD8+/CD44high T cells and Its Antitumor Action. J Immunol. 2008;180(4):2099–2106. doi:10.4049/jimmunol.180.4.2099.
- Epardaud M, Elpek KG, Rubinstein MP, Yonekura AR, Bellemare-Pelletier A, Bronson R, Hamerman JA, Goldrath AW, Turley SJ. Interleukin-15/interleukin-15R alpha complexes promote destruction of established tumors by reviving tumor-resident CD8+ T cells. Cancer Res. 2008;68: 2972–2983
- Pilones K, Aryankalayil J, Formenti S, Intratumoral DS. IL-15 potentiates radiation-induced anti-tumor immunity. Journal for Immunotherapy of Cancer. 2015;3(S2):P239 –P. doi:10.1186/2051-1426-3-S2-P239.
- Liu D, Song L, Wei J, Courtney AN, Gao X, Marinova E, Guo L, Heczey A, Asgharzadeh S, Kim E, et al. IL-15 protects NKT cells from inhibition by tumor-associated macrophages and enhances antimetastatic activity. J Clin Invest. 2012;122(6):2221–2233. doi:10.1172/JCI59535
- Nguyen R, Moustaki A, Norrie JL, Brown S, Akers WJ, Shirinifard A, Dyer MA. Interleukin-15 Enhances Anti-GD2 Antibody-Mediated Cytotoxicity in an Orthotopic PDX Model of Neuroblastoma. Clin Cancer Res. 2019;25(24):7554–7564. doi:10.1158/1078-0432.CCR-19-1045