
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Dendritic cell (DC) vaccination has proven to be an effective and safe adjuvant for cancer immunotherapies. As the presence of DCs within the tumor microenvironment promotes adaptive antitumor immunity, enhancement of DC migration toward the tumor microenvironment following DC vaccination might represent one possible approach to increase its therapeutic efficacy. While recent findings suggest the activity-regulated cytoskeleton-associated protein/activity-regulated gene 3.1 (Arc/Arg3.1) as critical regulator of DC migration in the context of autoimmune diseases, we aimed to investigate the impact of Arc/Arg3.1 expression for DC-based cancer vaccines.
To this end, DC migration capacity as well as the induction of T cell-mediated antitumor immunity was assessed in an experimental B16 melanoma model with Arc/Arg3.1−/- and Arc/Arg3.1-expressing BMDCs applied as a subcutaneous vaccine.
While antigen presentation on DCs was critical for unleashing effective T cell mediated antitumor immune responses, Arc/Arg3.1 expression enhanced DC migration toward the tumor and secondary lymphoid organs. Moreover, Arc/Arg3.1-expressing BMDCs shape the tumor immune microenvironment by facilitating tumor recruitment of antigen-specific effector T cells.
Thus, Arc/Arg3.1 may represent a novel therapeutic target in DCs in order to increase the therapeutic efficacy of DC vaccination.
Introduction
Dendritic cells (DC) are key mediators at the interface between host and adaptive immunity.Citation1 Localized in peripheral tissues, immature DCs patrol for invading pathogens. After capturing and processing antigens, DCs undergo maturation and migrate through lymphatic vessels to the site of antigen presentation and immune cell stimulation in the lymphoid organs. There, DCs present the captured antigens as processed MHC-bound peptides to effector T cells for the induction of T cell activation in order to initiate a T cell-based immune response against invading pathogens.Citation2
Preclinical proof-of-principle studies have shown that ex vivo generated and antigen-loaded DCs used as antitumor vaccines mount an antigen-specific T cell mediated immune response.Citation3,Citation4 Consistently, DC-based cancer vaccines also demonstrated therapeutic efficacy and a robust safety profile in clinical studies on different tumor entities.Citation5–8 However, DC vaccines as monotherapy could only hardly engender durable immune responses, pointing to the need for further approaches to enhance its therapeutic efficacy.Citation9
The current state of research implicates the application of DC vaccination in combination with other antitumor therapies.Citation10,Citation11 In this context, the combination of DC vaccination with adoptive T cell therapiesCitation12 lead to durable tumor control in preclinicalCitation13 and clinical studies.Citation14,Citation15
Further studies investigated important functions of distinct DC subpopulations in the tumor microenvironment.Citation16,Citation17 The murine classical DC1 (cDC1) subpopulation is characterized by CD8a and CD103 expression and depends on the transcription factors BATF3 and IRF8 during development.Citation18–20 CD103+ DCs take up tumor antigens and subsequently transport them CCR7-dependent to lymph nodes for further T cell priming.Citation21 CD8a+ DCs prime and activate CD8+ T cells by cross-presentation.Citation22 Recent investigations propose a crucial function of cDC1s in the tumor microenvironment for the induction of an effective T cell-based antitumor immune response.Citation23–25
The therapeutic efficacy of DC vaccination is critically dependent on the migration of subcutaneously injected DCs to their effector sites for T cell activation.Citation26–28 One possible approach to improve DC vaccination efficacy is to apply DCs with superior migratory capability for vaccination.Citation29 However, so far defined surrogate markers for DC migration are not exclusively expressed on migratory DCs and their expression levels vary between steady state and inflamed settings, hence cannot be widely used to select DCs with superior migratory capacity.Citation30,Citation31
Notably, we recently found that DCs with migratory capacity exclusively express the cytoskeleton-associated protein Arc/Arg3.1 and initiate T cell responses in inflammatory models.Citation32 Further ontogeny study showed that Arc/Arg3.1-expressing DCs are distributed among different DC subsets, including skin Langerhan cells (LC), cDC1 (CD103+) and cDC2 (CD11b+). Arc/Arg3.1 is expressed in 1–2% of in vitro generated, GM-CSF cultured BMDCs and in 10–40% of migratory DC subsets in vivo. The differentiation of Arc/Arg3.1-expressing DCs in vivo was found to be independent of specific transcription factors, suggesting Arc/Arg3.1 as an unequivocal functional marker for DCs with migratory capacity across all DC subsets.Citation33
These results directed us to the question whether the effect of Arc/Arg3.1 on DC migration may be translatable to immunotherapy for cancer treatment. In this study, we investigated the role of Arc/Arg3.1-dependent DC migration following DC vaccination for its therapeutic efficacy and capability to induce an antigen-specific T cell response in murine experimental melanoma.
Materials and methods
Mice
For in vivo tumor experiments, we used 7–9 weeks old male C57BL/6J Ly5.1 mice bred at the animal facility of the German Cancer Research Center Heidelberg or purchased from The Jackson Laboratory. Arc/Arg3.1-/- miceCitation34 were bred at the University Medical Center Hamburg-Eppendorf. Pmel-1 TCR transgenic mice [B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J] specific for the mouse homologue of human melanoma antigen hgp10025-33Citation35 were purchased from The Jackson Laboratory. Pmel-1/luc-mcherry mice were generated by crossing pmel-1 mice as above with luc-mcherry mice. Luc-mCherry mice, full name B6-Tg(Actb-Luc,mCherry)#Platt, express luciferase and mCherry under the Actb promotor and were generated in the Transgenic Service of the Center for Preclinical Research, DKFZ. All mice were bred under specific pathogen-free conditions. Animal experimental procedures were carried out according to institutional laboratory animal research guidelines and approved by the governmental authorities.
Cell culture
B16 wild type (WT) melanoma tumor cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 µg/ml streptomycin (all Sigma-Aldrich) at 37 °C, 5% CO2.
For tumor inoculation, cells were harvested with StemPro Accutase (Thermo Fischer, A1110501) and diluted in PBS (Sigma Aldrich) for injection.
Generation of murine CD8+ CTL
To obtain murine CD8+ CTL, spleens and lymph nodes of 6–10 weeks old pmel-1 or pmel-luc-mcherry mice were excised and meshed through a 70 µm cell strainer. After lysis of erythrocytes with ACK lysis buffer (150 mM NH4Cl, 10 mM KHCO3 and 100 μM Na2EDTA), the isolated immune cells were cultured in murine T cell proliferation medium consisting of RPMI-1640 (Sigma-Aldrich) supplemented with 10% FBS, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 25 mM Hepes pH 7.4, 1 mM sodium pyruvate, 5 × 10−5 M 2-mercaptoethanol (all Sigma-Aldrich) and 2 mM L-glutamine (Thermo-Fisher) under stimulation with 30 IU/ml IL-2 (Proleukin, Novartis) and 2 µg/ml hgp10025-33 (custom-made; Research Group GMP & T cell thrapy, DKFZ) for 3 days at 37 °C, 5% CO2. For adoptive cell transfer, CD8+ T cells were purified by using mouse CD8+ T cell isolation MACS Kit (MACS Miltenyi Biotec) according to the manufacturer’s instructions.
Generation of bone marrow derived DCs (BMDC)
Bone marrow was isolated from femurs and tibiae of 5–8 weeks old male C57BL/6J mice or Arc/Arg3.1−/–mice. After removal of remaining tissues from the bones, bone marrow was flushed out with PBS and homogenized with a 70 µm cell strainer. Bone marrow, if not otherwise given, was cultured in murine BMDC medium consisting of RPMI-1640 (Sigma-Aldrich) with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 25 mM Hepes pH 7.4, 1 mM sodium pyruvate, 5 × 10−5 M 2-mercaptoethanol (all Sigma-Aldrich) and 2 mM L-glutamine (Thermo-Fisher) in the presence of 20 ng/mL GM-CSF (Peprotech, 315–03) for 6 days at 37 °C, 5% CO2. We changed medium every other day by carefully replacing the supernatant with a fresh medium containing 20 ng/mL GM-CSF. We harvested non-adherent BMDCs on day six. For vaccination, BMDCs were matured with 100 ng/mL LPS (Sigma-Aldrich) for 24 h and, prior to vaccination, loaded for 4 h with 10 µg/ml hgp10025-33 or ovalbumin (OVA257-264) as control.Citation36
Production of pMXS-Arc/Arg3.1-IRES-GFP DNA construct
The Arc/Arg3.1-gene was cloned into the vector back bone pMXS-IRES-GFP using Gateway clonase enzymes according to manufacturer’s manual (Thermo Fischer). pMXS-IRES-GFP vector was a kind gift of Stefan Pusch (CCU Neuropathology, DKFZ Heidelberg).
Firstly, the full-length construct of Arc/Arg3.1 was flanked with attB-sites and cloned via Gateway BP reaction into the backbone of pDONR vector, from which the Arc/Arg3.1-gene was constitutively cloned into pMXS-IRES-GFP backbone vector via Gateway LR reaction.
Transduction of BMDCs
For the package of retrovirus, HEK Phoenix Eco I cells were transfected with the pMXS-Arc/Arg3.1-IRES-GFP construct (pMXS-Arc/Arg3.1) or the control vector pMXS-GFP-IRES-GFP (pMXS-control) using FuGENE® HD transfection reagent (Promega, E2311). Virus containing cell supernatant was harvested 48 hours after transfection for the subsequent transduction. Before retroviral transduction, freshly isolated bone marrow cells were pretreated with a cytokine cocktail mix consisting of 20 ng/mL IL-3, 50 ng/mL IL-6, 50 ng/mL SCF and 50 ng/mL TPO (all Peprotech) for 4 days.Citation37 20 ng/mL GM-CSF was added to the cell culture medium starting from day 2 after isolation of BMDCs. BMDCs were subsequently transduced at day 5 in RetroNectin (Takara) pre-coated 6-well plates with Polybrene of 10 µg/ml. Retrovirus containing supernatants were added to the cells, followed by centrifugation of the cell-virus mixtures at 1.200 g for 90 minutes to ensure contact between cells and virus particles. Next day, cells were washed with PBS to remove the remaining virus particles. The transduced BMDCs were maintained in cell culture for additional 4 days and stimulated with 100 ng/ml LPS (Sigma-Aldrich) for 24 h before harvest for vaccination. Transduction efficiency was examined via flow cytometric analysis for GFP+ cells.
Tumor challenge and treatment
For tumor inoculation, a cell suspension with 5 × 104 B16 wild type (WT) melanoma cells diluted in 100 µl PBS was mixed with 100 µl Matrigel Matrix (Corning) and injected into the right flank of the mice. To evaluate tumor growth, tumor area (width x length) was measured starting from day 6 after inoculation. For vaccination, 4 × 106 DCs were injected subcutaneously into the right hind leg at day 7 after tumor inoculation, followed by injection of 100.000 IU IL-2 per day (Proleukin, Novartis) for the two following days. A second vaccination with DCs was performed up to 6 days after the first vaccination.Citation13 For adoptive T cell transfer, 5 × 106 pmel-1 CD8+ T cells were intravenously injected at day 7 after inoculationCitation38 ().
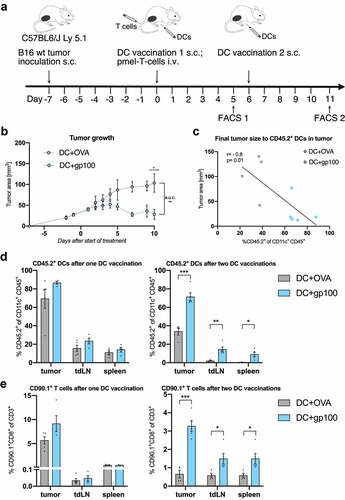
Figure 1. Vaccination with TAA-loaded BMDCs induces T cell mediated antitumor immune response (n=4-5 per group). (A) Scheme of B16 melanoma inoculation followed by combination treatment with DC vaccination and adoptive T cell transfer starting on day seven after tumor inoculation. Flow cytometry analysis was performed five days after first or second DC vaccination respectively. (B) Tumor growth curves of treated mice measured from two days before until ten days after start of treatment. (C) Correlation of final tumor sizes to tumor infiltrating CD45.2+CD11c+ DCs. (D-E) Flow cytometry analysis of frequencies of injected CD45.2+ DCs among all CD11c+ DCs (D) and CD90.1+CD8+ pmel T cells among all CD3+ T cells (E) in the tumor, tdLN and the spleen as measured five days after first or second vaccination. All data are presented as mean SEM. For (B), (D) and (E) we performed a two-tailed student’s t test to determine statistical significance (* p<0.05; ** p<0.01, *** p<0.001, **** p<0.0001). For (C) we calculated Pearson’s correlation coefficient to determine correlation
In vivo bioluminescent imaging
For in vivo bioluminescent imaging, we used the IVIS Lumina Series III from Perkin Elmer. Before imaging, mice were shaved at the body regions of interest, injected with 50 mg/kg D-Luciferin i.p. (StayBrite™, BioVision, Mountain View) and anesthesia was induced with isoflurane 3–4%. Bioluminescence images were acquired 10 minutes after D-Luciferin injection with an exposure time of 30, 60 and 90 seconds. During imaging, mice were kept under anesthesia with isoflurane 1,5%.
Quantification of bioluminescence signals was performed by the Living Image 4.3 Software (Perkin Elmer). Therefore, regions of interest were drawn around the regions of the tumor and secondary lymphoid organs () and signals were quantified as photons/second.
Isolation of leukocytes from tumor, blood, lymph nodes and spleen
For the isolation of tumor infiltrating lymphocytes, mice were killed by terminal cardial perfusion with 20 ml PBS nine to eleven days after start of treatment. Whole tumor tissues were excised and digested with HBSS supplemented with 0,5 mg/ml Collagenase D (Roche) and 20 µg/ml DNAse I (Sigma-Aldrich) at 37 °C for 60 minutes. Digested tumors were meshed twice through a 100 µm and a 70 µm cell strainer to obtain single-cell suspension. Erythrocytes were lysed with ACK Lysis Buffer. For the isolation of splenocyte, spleens were excised and meshed twice through a 70 µm cell strainer, followed by lysis of erythrocytes in ACK Lysis Buffer. Tumor draining lymph nodes were excised from the inguinal and axillary region of the tumor bearing side (right side) of the mouse. For further processing, lymph nodes were meshed through a 70 µm cell strainer to obtain a single-cell suspension.
Flow cytometry
Flow cytometry analyses of immune cell subsets ex vivo were performed 5 days following the last DC vaccination.
Single-cell suspensions from tumor, spleen and lymph nodes samples were washed thoroughly for cell staining and blocked with anti-CD16/32 (Biolegend) before staining. For extracellular targets, cells were stained for 30 minutes at 4 °C.
For intracellular staining, cells were incubated with 5 µg/mL Brefeldin A (Sigma Aldrich) at 37 °C, 5% CO2 for 5 h. For IFNγ detection, cells were restimulated with 10 mg/ml hgp10025-33 at 37 °C, 5% CO2 for 5 h. Intracellular cell staining was performed for 45 min at 4 °C.
Flow cytometry was measured by using BD FACS Canto II or Attune NxT analyzers.
Data were analyzed with FlowJo V10.
For the characterization of in vitro generated murine BMDCs, a single-cell suspension of 1–3*106 cells were stained with following extracellular antibodies: fixable viability dye – eFluor 780 (Thermo Fischer, 65086514); anti-CD11c-APC (Biolegend, clone N418, 117310); anti-I-A/I-E-BV711(Biolegend, clone M5/114.15.2, 107643).
For ex vivo identification of cytotoxic T cells from the tumor and secondary lymphoid organs, we used following antibodies: fixable viability dye – APC-Cy7/eFluor 780 (Thermo Fischer, 65086514); anti-CD45-BV510 (Biolegend, clone 30-F11, 103138); anti-CD3-BV711 (Biolegend, clone 17A2, 100241); anti-CD8-AF700 (Biolegend, clone 53–6.7, 100730); anti-CD4-PE-Texas Red (Thermo Fischer, clone RM4-5, MCD0417); anti-CD90.1-PE (Biolegend, clone OX-7, 202524). T-cells were identified by gating for CD45+CD3+ population on CD45+ live cells. From there, further gating was performed on CD8+ for cytotoxic T-cells and CD90.1+ for pmel-1 gp100 specific T-cells.
For ex vivo identification of DCs from the tumor and secondary lymphoid organs, the single-cell suspension was stained using following extracellular antibodies: fixable viability dye – eFluor 780 (Thermo Fischer, 65086514); anti-CD45-BV510 (Biolegend, clone 30-F11, 103137); anti-CD45.2-eFluor 450 (Thermo-Fischer, clone 104, 48045482); anti-CD11c-APC (Biolegend, clone N418, 117310); anti-I-A/I-E-BV711(Biolegend, clone M5/114.15.2, 107643); anti-CD8a-PerCP-Cy5.5 (Thermo Fischer, clone 53–6.7, 45008182) and anti-CD103-PE (Biolegend, clone 2E7, 121406). Dendritic cells were identified as CD11c+ from CD45+ live cells. Injected DCs were gated as CD45.2+CD11c+ cells (Fig. S1A) and so distinguished from recipient mice intrinsic dendritic cells. For further phenotypic characterization, identified dendritic cells were gated for CD8a+, CD103+ and MHCII+ cell population.
Immunoblot
We performed immunoblotting on whole-cell lysates. 40 µg of protein were subjected to SDS–polyacrylamide gel electrophoresis and transferred to nitrocellulose. After blocking, we incubated the membranes with antibodies directed to Arc/Arg3.1 (mouse, 1:4000; Worley Lab) as previously describedCitation39 or H3 (rabbit, 1:1000; Cell Signaling, cat. 9715) overnight at 4 °C and washed and incubated them with a species-specific secondary antibody (1:20000; LI-COR Biosciences) for 1 hour at room temperature. Labeling was visualized using enhanced chemiluminescence (LI-COR Biosciences). Quantification was carried out by densitometry using ImageJ software. For uncropped immunoblots, see Fig. S2B.
mRNA sequencing
Purification of RNA from FACS sorted migratory DCs from sdLN of C57BL/6 J mice was done with the RNeasy Mini (Qiagen) according to the manufacturer’s protocol. RNA-seq libraries were prepared using the NEBNext Ultra RNA Library Prep Kit for Illumina (New England Biolabs) and sequenced on an HiSeq 2000 sequencer (Illumina) generating 50 base pair single-end reads. The reads were aligned to the Ensembl mouse reference genome (mm10) using STAR v.2.4. The overlap with annotated gene loci was counted with featureCounts v.1.5.1.
All analyses were performed in the R environment (v.4.0.4) using publicly available packages.
Statistical analysis
Data analyses were performed with GraphPad Prism 8, if not stated otherwise. All data are presented as means SEM as indicated in figure legends. Data were analyzed by two-tailed Student’s t-test for comparison of two groups and one-way ANOVA combined with correction for multiple testing for comparison of three groups. Correlation was determined by calculation of Pearson’s correlation coefficient. p < .05 was considered statistically significant (* p < .05; ** p < .01, *** p < .001, **** p < .0001).
Data availability
Data generated for this study are available from the correspondent author upon reasonable request.
Results
TAA-loaded BMDCs in the tumor microenvironment induce T cell mediated antitumor immune response
Therapeutic efficacy of a tumor antigen-specific DC vaccine was assessed by application of BMDCs loaded with the tumor-associated antigen hgp10025-33 in combination with gp100-specific pmel-1 T cells to B16 melanoma bearing mice. For comparison, BMDCs loaded with OVA257-264 were injected as an unspecific control vaccination (). According to clinical application in melanoma patients, DC vaccine was applied subcutaneously.Citation40
TAA-loading of DCs determined response to adoptive T cell therapy, since tumor growth control was only achieved following tumor-antigen specific DC vaccination (). Of note, higher frequencies of tumor infiltrating CD45.2+ donor DCs were associated with smaller tumor sizes (, Fig. S1A). A boost vaccination with TAA-loaded BMDCs significantly increased the frequencies of CD45.2+ DCs within the tumor tissue and the secondary lymphoid organs in comparison to the control boost vaccination with OVA-loaded BMDCs (). In line, a second vaccination with TAA-loaded BMDCs was associated with an increased recruitment of tumor-specific CD90.1+ T cells toward the tumor tissue and secondary lymphoid organs at day 18 after tumor injection ().
Arc/Arg3.1 expression is crucial for migration of donor BMDCs following DC vaccination
Our previous findings suggest a crucial role of DC migration toward tumors and secondary lymphoid organs for mounting an effective T cell mediated antitumor immune response. To determine the relevance of Arc/Arg3.1 expression in DCs for an antitumor immune response, therapeutic efficacy of DC vaccination using BMDCs isolated from Arc/Arg3.1-deficient (Arc/Arg3.1-/-) miceCitation34 and Arc/Arg3.1-expressing wild-type mice (WT, Arc/Arg3.1+/+) was explored.
Migration of CD45.2+ donor DCs to the tumor () and to the secondary lymphoid organs () was reduced after vaccination with TAA-loaded Arc/Arg3.1−/–BMDCs in comparison to Arc/Arg3.1+/+-BMDCs.
Figure 2. Arc/Arg3.1-dependent migration of injected TAA-loaded BMDCs to the tumor and secondary lymphoid organs following DC vaccination (n=4-5 per group). Flow cytometry analysis of injected CD45.2+ DCs eleven days after start of treatment. (A-C) Frequency of injected CD45.2+ DCs among all CD11c+ DCs in the tumor (A), tdLN (B) and the spleen (C). (D) Expression of DC activation markers MHCII and CD86 on tumor infiltrating CD45.2+. (E) Frequency of donor-derived CD45.2+ CD8a+ DCs among all CD11c+ DCs in the tumor and respective gating strategy for CD45.2+ CD8a+ DCs. All data are presented as mean SEM. We performed a one-way ANOVA in combination with Tukeys’s test to determine statistical significance (* p<0.05; ** p<0.01, *** p<0.001)
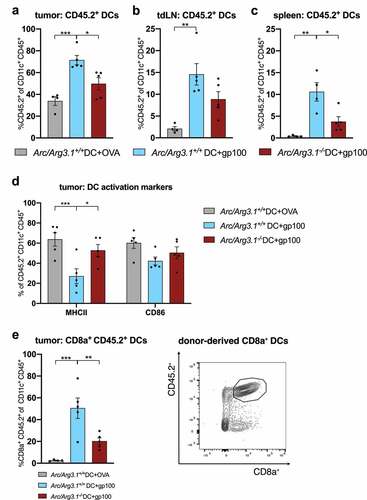
In line with previous findings, Citation32 Arc/Arg3.1 deficiency did not impair in vitro BMDC maturation and activation, respectively (Fig. S1B). In vivo, Arc/Arg3.1 deficiency was associated with an increased MHC class II expression on donor-derived TAA-loaded DCs in the tumor (). cDC subtypes including CD8a+ and CD103+ cDC1 and CD11b+ cDC2 were represented among tumor infiltrating CD45.2+ DCs (Fig. S1C). Notably, the frequency of CD8a+ DCs among all CD11c+ donor DCs in the TME depended on both TAA-loading and Arc/Arg3.1 expression on injected BMDCs ().
Collectively, our findings imply that Arc/Arg3.1 is critical for migration of subcutaneously injected TAA-loaded DCs to the tumor and secondary lymphoid organs.
Vaccination with Arc/Arg3.1-expressing BMDCs promotes recruitment of activated TAA-specific T cells to the tumor microenvironment
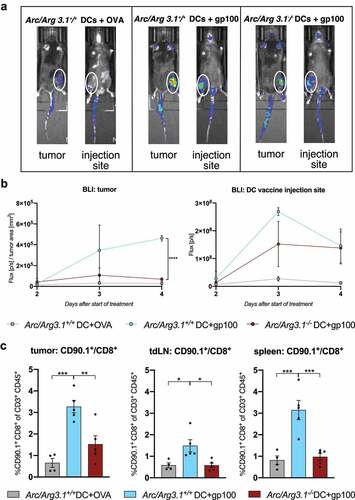
We next investigated the role of Arc/Arg3.1 expression in DC vaccines for the recruitment of tumor-specific T cells. Luminescence signals from adoptively transferred luciferase-expressing CD8+ effector T cells (pmel luc mcherry)Citation41 at the tumor site as well as at the DC vaccine injection site were monitored (). While TAA-loading of donor DCs was critical for T cell recruitment to the vaccine injection site, tumor trafficking of antigen-specific T cells was dependent on Arc/Arg3.1 expression of DCs: Four days after adoptive transfer, tumor infiltration of gp100-specific CD8+ T cells was significantly enhanced after vaccination with gp100-loaded WT-BMDCs as compared to gp100-loaded Arc/Arg3.1−/–BMDCs or OVA-loaded WT-BMDCs (). Luminescence signal at the tumor site increased starting from day two until day four after adoptive transfer and decreased at the DC vaccine injection site starting on day three after treatment indicating a migration of tumor-specific CD8+ effector T cells from the site of DC vaccination to the tumor site (). In line, flow cytometry analyses revealed an increased infiltration of CD90.1+ T cells to the tumor and secondary lymphoid organs following vaccination with Arc/Arg3.1-expressing BMDCs in comparison to the controls () and expression profiling of Arc/Arg3.1-expressing migratory DCs revealed expression of T cell recruiting chemokines (Fig. S1D).
Figure 3. Recruitment of antigen-specific T cells to the tumor microenvironment depends on Arc/Arg3.1 expression in injected BMDCs. (A-B) In vivo bioluminescence imaging (IVIS) of adoptively transferred pmel luc mcherry T cells two to four days after start of treatment. (A) Photographic images of bioluminescence signals from luciferase expressing T cells in vivo in the tumor and at the DC vaccine injection site four days after start of treatment. (B) Quantification of bioluminescence signals from luciferase expressing T cells in the tumor and at the DC vaccine injection site measured from two days until four days after start of treatment. Signal is measured in photon/s and normalized to tumor size at respective days of measurements (n=3). (C) Flow cytometry analysis of frequency of CD90.1+CD8+ pmel T cells among all CD3+ T cells in the tumor, tdLN and the spleen eleven days after start of treatment (n=4-5 per group). All data are presented as mean SEM. For (B)-(C) we performed a one-way ANOVA in combination with Tukeys’s test to determine statistical significance (* p<0.05; ** p<0.01, *** p<0.001)
Arc/Arg3.1-overexpressing BMDCs in the tumor and secondary lymphoid organs following DC vaccination
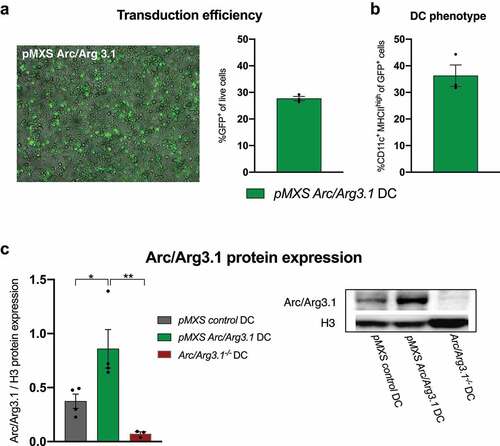
To further investigate the effect of Arc/Arg3.1 expression for DC vaccination, Arc/Arg3.1-overexpressing BMDCs were generated.Citation42 Following transduction of WT BMDCs with the retroviral construct pMXS-Arc/Arg3.1-IRES-GFP (pMXS-Arc/Arg3.1), transduction efficiency with 30% GFP+ cells was quantified by flow cytometry (, S2A) and DC phenotype was confirmed by CD11c and MHCII expression (, S2A).Citation43 Immunoblot verified Arc/Arg3.1 overexpression in pMXS-Arc/Arg3.1-transduced DCs (, S2B).
Figure 4. Retroviral transduction of BMDCs for the overexpression of Arc/Arg3.1. (A) Fluorescence microscopy of GFP and flow cytometry analysis of GFP+ cells after retroviral transduction with pMXS-Arc/Arg3.1-IRES-GFP (n=3). (B) Flow cytometry analysis of CD11c+ MHCII+ BMDCs among GFP+ cells after retroviral transduction with pMXS-Arc/Arg3.1-IRES-GFP (n=3). (C) Immunoblot of Arc/Arg3.1 from pMXS-control transduced, pMXS-Arc/Arg3.1 transduced and Arc/Arg3.1-/--BMDCs (n=3-4 per group). All data are presented as mean SEM. For (C) we performed a one-way ANOVA in combination with Tukeys’s test to determine statistical significance (* p<0.05; ** p<0.01)
Following vaccination of B16 melanoma bearing mice with TAA-loaded Arc/Arg3.1-overexpressing DCs, donor DCs could be detected in the tumor and secondary lymphoid organs based on their GFP expression (). Hereby, increased Arc/Arg3.1 expression enhanced DC migration to the tdLN as demonstrated by a positive correlation between MFI GFP representing pMXS-Arc/Arg3.1 expression and the frequency of CD45.2+ donor DCs in the tdLN (). This positive correlation could not be detected when applying pMXS-control transduced BMDCs (). Arc/Arg3.1 overexpression of BMDCs did not impair DC activation as assessed by MHC II expression (Fig. S3A) nor influence cDC subpopulation frequencies within the tumor (Fig. S3B). Even so, an increase in Arc/Arg3.1 expression did not provide a therapeutic benefit on tumor growth following DC vaccination (Fig. S3C, D).
Figure 5. Arc/Arg3.1-overexpressing BMDCs in the tumor and secondary lymphoid organs following DC vaccination. Flow cytometry analysis of CD11c+ DCs in the tumor nine days after start of treatment. (A) Quantification of GFP signal (MFI) in flow cytometry on injected CD45.2+CD11c+ BMDCs and endogenous CD45.2-CD11c+ BMDCs in the tumor, tdLN and the spleen (n=11). (B) Correlation of donor derived CD45.2+CD11c+ DCs to MFI of GFP on CD45.2+ CD11c+ DCs in tdLN (n=11). All data are presented as mean SEM. For (A) we used a two-tailed student’s t test to determine statistical significance (* p<0.05; ** p<0.01, *** p<0.001, **** p<0.0001). For (B), Pearson’s correlation coefficient was calculated to determine correlation
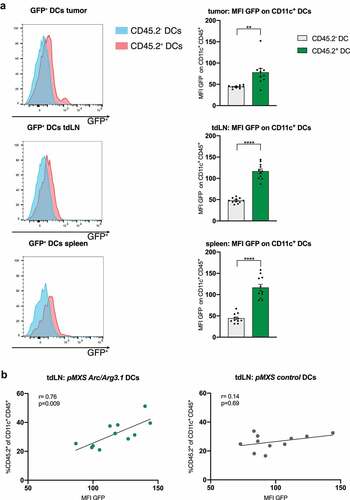
Arc/Arg3.1-overexpressing BMDCs shape the tumor immune microenvironment
Correlation analyses of CD45.2+ donor DCs to the presence of CD90.1+ T cells within the TME revealed a significant positive correlation between antigen-specific T cell infiltration and the infiltration of injected BMDCs in the tumor () and tdLN () exclusively after vaccination with Arc/Arg3.1-overexpressing DCs. However, Arc/Arg3.1 overexpression was not sufficient to increase activation and proliferation of prior in vitro stimulated antigen-specific T cells in the tumor compared to control gp100-loaded DCs ().
Figure 6. Antigen-specific T cells in the tumor microenvironment following vaccination with Arc/Arg3.1-overexpressing BMDCs. Flow cytometry analysis of CD45.2+CD11c+ DCs and CD90.1+CD8+ T cells in the tumor and tumor draining lymph nodes. (A)-(B) Correlation of CD90.1+CD8+ T cells to donor-derived CD45.2+CD11c+ DCs in the tumor (A) and tdLN (B) (n=17). (C) Expression of T cell activation and proliferation markers on tumor infiltrating CD90.1+CD8+ T cells. All data are presented as mean SEM. For (A) and (B) Pearson’s correlation coefficient was calculated to determine correlation
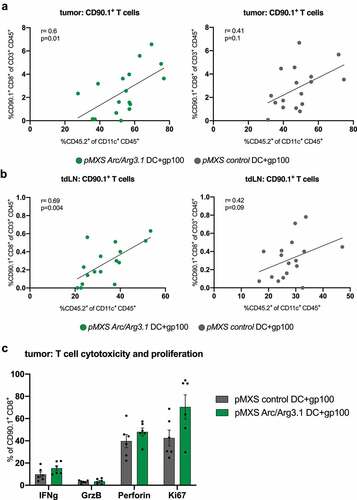
In summary, our results demonstrate that Arc/Arg3.1-expressing DCs in the TME are crucial for intratumoral accumulation of antigen-specific T cells.
Discussion
Clinical implications suggest the usage of DC vaccination in combination with an adoptive transfer of cytotoxic T cells as immunotherapy against cancer.Citation14 However, durable antitumor immune responses and therapeutic tumor control following DC vaccination are still limited and need to be further improved.Citation6
DC vaccination with BMDCs loaded with the TAA gp100 was therapeutically effective against gp100-expressing B16 melanoma (). Activation of gp100-specific pmel-1 T cells by gp100-loaded DCs in the TME enabled further recruitment of injected DCs, as observed by a higher frequency of donor-derived BMDCs in the tumor and secondary lymphoid organs following two sequential vaccinations with gp100-loaded BMDCs as compared to the control ().Citation13,Citation44 Since an increased tumor infiltration of injected DCs was associated with therapeutic response (), we developed the rationale to improve DC migration to the TME to enhance the therapeutic efficacy of DC vaccination.Citation29,Citation45 Of note, gp100 is also expressed in healthy skin; thus, autoimmune destruction of melanocytes resulting in vitiligo should be considered for clinical application.Citation35,Citation46 Yet, such side effects were not observed within this study.
Arc/Arg3.1 expression in DCs was critical for the migration of subcutaneously injected peptide-loaded BMDCs () and led to an increase of CD45.2+ CD8a+ DCs in the TME (). Previous observations demonstrated that Arc/Arg3.1 expression is not stimulated by DC migration promoting chemokines CCL19/CCL21 and that CCR7 expression on DCs was not impaired by Arc/Arg3.1-deficiency.Citation32 The reduced migration of Arc/Arg3.1−/–DCs to the tumor as well as to the secondary lymphoid organs () further indicates a chemokine-independent migration of LPS-activated Arc/Arg3.1-expressing DCs.
In the tumor microenvironment, resident Batf3-dependent cDC1s initiate effective T cell recruitment by secretion of T cell recruiting chemokines CXCL9/CXCL10,Citation47 which directed us to further investigate the antigen-specific T cell response following DC vaccination with Arc/Arg3.1-expressing BMDCs. Indeed, T cell trafficking to the TME was dependent on Arc/Arg3.1 expression in injected DCs (). Moreover, an increased frequency of adoptively transferred gp100-specific T cells was associated with an increase in donor derived gp100-loaded DCs in the tumor and tdLN exclusively after injecting Arc/Arg3.1-overexpressing DCs (). The expression of T cell recruiting chemokines by Arc/Arg3.1-expressing migratory DCs (Fig. S1D) supports the hypothesis that Arc/Arg3.1-expressing DCs in the TME are involved in the recruitment of antigen-specific T cells. In the lymphoid organs, resident CD8a+ cDC1s are essential for cross-presentation of tumor-derived antigens to T cells leading to T cell proliferation and activation.Citation48,Citation49 Also, cross presentation by a distinct Batf3-dependent DC subpopulation within the tumor microenvironment itself mediated T cell activation and determined tumor rejection.Citation25 It remains to be elucidated, whether processing of tumor antigens by injected donor-derived DCs for cross presentation to CD8+ cytotoxic T cells might be a reason for the observed decrease of MHC class II expression on donor-derived gp100-loaded DCs in the tumor ().Citation50
The migratory capacity of Arc/Arg3.1-expressing DCs serves as a potential target to enhance the therapeutic efficacy of DC vaccination by increasing the amount of Arc/Arg3.1-expressing DCs among in vitro generated BMDCs.
However, Arc/Arg3.1 overexpression in genetic-modified BMDCs was not sufficient to further enhance the therapeutic effect of DC vaccination on tumor growth as compared to vaccination with control-vector transduced BMDCs (Fig. S3C, D). The increase in Arc/Arg3.1-expressing DCs to 30% pMXS-Arc/Arg3.1+ DCs among all generated BMDCs following genetic modification () might be scarce to engender a therapeutic benefit for DC vaccination and thus limits its clinical application as antitumor therapy. Therapeutic efficacy might be augmented by an increase in transduction efficiency or the injection of purified Arc/Arg3.1-expressing DCs for vaccination. Although vaccination with Arc/Arg3.1-expressing BMDCs was associated with increased T cell infiltration to the tumor, the T cell mediated antitumor response might be limited by T cell exhaustion in the tumor micromilieu. Since the presence of Arc/Arg3.1-overexpressing DCs in the TME does not alter the cytotoxic potential of activated tumor-specific T cells, the vaccination with Arc/Arg3.1-expressing DCs might not be sufficient to unleash an effective T cell response in the tumor. A combination with immune checkpoint blocking antibodies could be evaluated to increase the therapeutic effect of Arc/Arg3.1-expressing DCs as antitumor therapy.
Our study depicts the role of Arc/Arg3.1-dependent DC migration to the tumor and lymphoid organs for the trafficking of antigen-specific T cells as essential part of an effective antitumor immune response after DC vaccination. T cell infiltration to the tumor was significantly reduced following DC vaccination when DC migration was impaired by deficient Arc/Arg3.1 expression in injected BMDCs () and an enhancement of Arc/Arg3.1 expression in injected BMDCs also significantly increased T cell infiltration to the tumor (). The usage of DCs with superior migratory capability for DC vaccination might serve as a potential therapeutic approach to enhance T cell mediated immune response against the tumor.
Authors contributions
X.W.Z., K.S. and M.P. conceptualized the studies and designed the experiments. X.W.Z., K.H. and F.C. developed methodologies and performed research and experiments. K.J., J.K.S., S.B., M.S.W., K.L. and M.K. performed in vitro, in vivo and ex vivo experiments. E.G., F.U. and M.A.F. provided expertise and helped developing methodologies. X.W.Z., K.H., J.K.S., F.U., M.A.F., M.P. and K.S. wrote the manuscript with input from all co-authors.
Disclosure of potential conflicts of interest
The authors declare no competing interests.
Supplemental Material
Download ()Acknowledgements
We acknowledge the support by the Center for Preclinical Research, the Core Facility for Flow Cytometry and for Light Microscopy at the German Cancer Research Center and the Flow Cytometry Core Facility at the Medical Faculty Mannheim of the Heidelberg University. We thank Paul Worley, Baltimore, USA, for providing anti-Arc/Arg3.1 antibody and Dietmar Kuhl, Hamburg, Germany for providing Arc/Arg3.1–/– mice.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245–13. doi:10.1038/32588.
- Stoll S, Delon J, Brotz TM, Germain RN. Dynamic imaging of t cell-dendritic cell interactions in lymph nodes. Science. 2002;296(5574):1873–1876. doi:10.1126/science.1071065
- Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg G, Schadendorf D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med. 1998;4(3):328–332. doi:10.1038/nm0398-328.
- Mayordomo JI, Zorina T, Storkus WJ, Zitvogel L, Celluzzi C, Falo LD, Melief CJ, Ildstad ST, Martin Kast W, Deleo AB, et al. Bone marrow-derived dendritic cells pulsed with synthetic tumour peptides elicit protective and therapeutic antitumour immunity. Nat Med. 1995;1(12):1297–1302. doi:10.1038/nm1295-1297.
- Nestle FO, Farkas A, Conrad C. Dendritic-cell-based therapeutic vaccination against cancer. Curr Opin Immunol. 2005;17:163–169. doi:10.1016/j.coi.2005.02.003.
- Anguille S, Smits EL, Lion E, Van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014;15(7):e257–67. doi:10.1016/S1470-2045(13)70585-0.
- Leonhartsberger N, Ramoner R, Falkensammer C, Rahm A, Gander H, Höltl L, Thurnher M. Quality of life during dendritic cell vaccination against metastatic renal cell carcinoma. Cancer Immunol Immunother CII. 2012;61(9):1407–1413. doi:10.1007/s00262-012-1207-7.
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. doi:10.1056/NEJMoa1001294.
- Boudewijns S, Bloemendal M, Gerritsen WR, De Vries IJM, Schreibelt G. Dendritic cell vaccination in melanoma patients: from promising results to future perspectives. Hum Vaccines Immunother. 2016;12(10):2523–2528. doi:10.1080/21645515.2016.1197453.
- Van Gulijk M, Dammeijer F, Aerts JGJV, Vroman H. Combination strategies to optimize efficacy of dendritic cell-based immunotherapy. Front Immunol. 2018:9. doi:10.3389/fimmu.2018.02759.
- Galati D, Zanotta S. Empowering dendritic cell cancer vaccination: the role of combinatorial strategies. Cytotherapy. 2018;20:1309–1323. doi:10.1016/j.jcyt.2018.09.007.
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi:10.1126/science.aaa4967.
- Lou Y, Wang G, Lizée G, Kim GJ, Finkelstein SE, Feng C, Restifo NP, Hwu P. Dendritic cells strongly boost the antitumor activity of adoptively transferred T cells in vivo. Cancer Res. 2004;64:6783–6790. doi:10.1158/0008-5472.CAN-04-1621
- Poschke I, Lövgren T, Adamson L, Nyström M, Andersson E, Hansson J, Tell R, Masucci GV, Kiessling R. A phase I clinical trial combining dendritic cell vaccination with adoptive T cell transfer in patients with stage IV melanoma. Cancer Immunol Immunother. 2014;63:1061–1071. doi:10.1007/s00262-014-1575-2.
- Kandalaft LE, Powell DJ, Chiang CL, Tanyi J, Kim S, Bosch M, Montone K, Mick R, Levine BL, Torigian DA, et al. Autologous lysate-pulsed dendritic cell vaccination followed by adoptive transfer of vaccine-primed ex vivo co-stimulated T cells in recurrent ovarian cancer. Oncoimmunology. 2013;2(1):e22664. doi:10.4161/onci.22664.
- Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2019 August 29;20(1):7–24. Published Online First. doi:10.1038/s41577-019-0210-z.
- Laoui D, Keirsse J, Morias Y, Van Overmeire E, Geeraerts X, Elkrim Y, Kiss M, Bolli E, Lahmar Q, Sichien D, et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat Commun. 2016;7(1):13720. doi:10.1038/ncomms13720.
- Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, Yona S. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. 2014;14(8):571–578. doi:10.1038/nri3712.
- Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31(1):563–604. doi:10.1146/annurev-immunol-020711-074950.
- Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, Moon C, Albring JC, Ise W, Michael DG, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med. 2010;207(4):823–836. doi:10.1084/jem.20091627.
- Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, Krummel MF, et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of t cell immunity in melanoma. Cancer Cell. 2016;30(2):324–336. doi:10.1016/j.ccell.2016.06.003.
- Den Haan JM, Lehar SM, Bevan MJ. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med. 2000;192:1685–1696. doi:10.1084/jem.192.12.1685.
- Böttcher JP, Sousa CRE. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer. 2018;4:784–792. doi:10.1016/j.trecan.2018.09.001.
- Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, Rogers NC, Sahai E, Zelenay S, Reis E Sousa C, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172(5):1022–1037. e14. doi:10.1016/j.cell.2018.01.004.
- Broz ML, Binnewies M, Boldajipour B, Nelson A, Pollack J, Erle D, Barczak A, Rosenblum M, Daud A, Barber D, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T Cell immunity. Cancer Cell. 2014;26(5):638–652. doi:10.1016/j.ccell.2014.09.007.
- Lappin MB, Weiss JM, Delattre V, Mai B, Dittmar H, Maier C, Manke K, Grabbe S, Martin S, Simon JC. Analysis of mouse dendritic cell migration in vivo upon subcutaneous and intravenous injection. Immunology. 1999;98(2):181–8. doi:10.1046/j.1365-2567.1999.00850.x
- Verdijk P, Aarntzen EHJG, Lesterhuis WJ, Boullart ACI, Kok E, Van Rossum MM, Strijk S, Eijckeler F, Bonenkamp JJ, Jacobs JFM, et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin Cancer Res Off J Am Assoc Cancer Res. 2009;15(7):2531–2540. doi:10.1158/1078-0432.CCR-08-2729.
- Eggert AAO, Schreurs MWJ, Boerman OC, Oyen WJ, De Boer AJ, Punt CJ, Figdor CG, Adema GJ. Biodistribution and vaccine efficiency of murine dendritic cells are dependent on the route of administration. Cancer Res. 1999;59:3340–3345.
- Okada N, Mori N, Koretomo R, Okada Y, Nakayama T, Yoshie O, Mizuguchi H, Hayakawa T, Nakagawa S, Mayumi T, et al. Augmentation of the migratory ability of DC-based vaccine into regional lymph nodes by efficient CCR7 gene transduction. Gene Ther. 2005;12(2):129–139. doi:10.1038/sj.gt.3302358.
- Worbs T, Hammerschmidt SI, Förster R. Dendritic cell migration in health and disease. Nat Rev Immunol. 2017;17:30–48. doi:10.1038/nri.2016.116.
- Helft J, Ginhoux F, Bogunovic M, Merad M. Origin and functional heterogeneity of non-lymphoid tissue dendritic cells in mice. Immunol Rev. 2010;234(1):55–75. doi:10.1111/j.0105-2896.2009.00885.x.
- Ufer F, Vargas P, Engler JB, Tintelnot J, Schattling B, Winkler H, Bauer S, Kursawe N, Willing A, Keminer O, et al. Arc/Arg3.1 governs inflammatory dendritic cell migration from the skin and thereby controls T cell activation. Sci Immunol. 2016;1(3):eaaf8665. doi:10.1126/sciimmunol.aaf8665.
- Tintelnot J, Ufer F, Engler JB, Winkler H, Lücke K, Mittrücker H-W, Friese MA. Arc/Arg3.1 defines dendritic cells and Langerhans cells with superior migratory ability independent of phenotype and ontogeny in mice. Eur J Immunol. 2019;49(5):724–736. doi:10.1002/eji.201847797.
- Plath N, Ohana O, Dammermann B, Errington ML, Schmitz D, Gross C, Mao X, Engelsberg A, Mahlke C, Welzl H, et al. Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron. 2006;52(3):437–444. doi:10.1016/j.neuron.2006.08.024.
- Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, De Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T Cells. J Exp Med. 2003;198(4):569–580. doi:10.1084/jem.20030590.
- Obermajer N, Urban J, Wieckowski E, Muthuswamy R, Ravindranathan R, Bartlett DL, Kalinski P. Promoting the accumulation of tumor-specific T cells in tumor tissues by dendritic cell vaccines and chemokine-modulating agents. Nat Protoc. 2018;13:335–357. doi:10.1038/nprot.2017.130
- Li G-B, Lu G-X. Gene delivery efficiency in bone marrow-derived dendritic cells: comparison of four methods and optimization for lentivirus transduction. Mol Biotechnol. 2009;43:250–256. doi:10.1007/s12033-009-9197-1.
- Abad JD, Wrzensinski C, Overwijk W, De Witte MA, Jorritsma A, Hsu G, Gattinoni L, Cohen CJ, Paulos CM, Palmer DC et al. T-Cell Receptor Gene Therapy of Established Tumors in a Murine Melanoma Model. J Immunother Hagerstown Md 1997 2008;31:1–6. doi:10.1097/CJI.0b013e31815c193f
- Alberi L, Liu S, Wang Y, Badie R, Smith-Hicks C, Wu J, Pierfelice TJ, Abazyan B, Mattson MP, Kuhl D, et al. Activity-induced notch signaling in neurons requires Arc/Arg3.1 and is essential for synaptic plasticity in hippocampal networks. Neuron. 2011;69(3):437–444. doi:10.1016/j.neuron.2011.01.004.
- Lesterhuis WJ, Vries IJMD, Schreibelt G, Lambeck AJA, Aarntzen EHJG, Jacobs JFM, Scharenborg NM, Van De Rakt MWMM, De Boer AJ, Croockewit S, et al. Route of administration modulates the induction of dendritic cell vaccine–induced antigen-specific T Cells in advanced melanoma patients. Clin Cancer Res. 2011;17(17):5725–5735. doi:10.1158/1078-0432.CCR-11-1261.
- Palmer DC, Balasubramaniam S, Hanada K, Wrzesinski C, Yu Z, Farid S, Theoret MR, Hwang LN, Klebanoff CA, Gattinoni L et al. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumor destruction. J Immunol Baltim Md 1950. 2004;173: 7209–16
- Abraham RS, Mitchell DA. Gene-modified dendritic cell vaccines for cancer. Cytotherapy. 2016;18(11):1446–1455. doi:10.1016/j.jcyt.2016.09.009.
- Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml B, Goubau D, Reis e sousa C. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity. 2015;42(6):1197–1211. doi:10.1016/j.immuni.2015.05.018.
- Nguyen-Pham T-N, Lim M-S, Nguyen TAT, Lee Y-K, Jin C-J, Lee HJ, Hong CY, Ahn J-S, Yang D-H, Kim Y-K, et al. Type I and II interferons enhance dendritic cell maturation and migration capacity by regulating CD38 and CD74 that have synergistic effects with TLR agonists. Cell Mol Immunol. 2011;8(4):341–347. doi:10.1038/cmi.2011.7.
- Seyfizadeh N, Muthuswamy R, Mitchell DA, Nierkens S, Seyfizadeh N. Migration of dendritic cells to the lymph nodes and its enhancement to drive anti-tumor responses. Crit Rev Oncol Hematol. 2016;107:100–110. doi:10.1016/j.critrevonc.2016.09.002.
- Luiten RM, Kueter EWM, Mooi W, Gallee MPW, Rankin EM, Gerritsen WR, Clift SM, Nooijen WJ, Weder P, Van De Kasteele WF, et al. Immunogenicity, including vitiligo, and feasibility of vaccination with autologous GM-CSF–transduced tumor cells in metastatic melanoma patients. J Clin Oncol Off J Am Soc Clin Oncol. 2005;23(35):8978–8991. doi:10.1200/JCO.2005.01.6816.
- Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31(5):711–723. e4. doi:10.1016/j.ccell.2017.04.003.
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322(5904):1097–1100. doi:10.1126/science.1164206.
- Sánchez-Paulete AR, Teijeira A, Cueto FJ, Garasa S, Pérez-Gracia JL, Sánchez-Arráez A, Sancho D, Melero I. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann Oncol Off J Eur Soc Med Oncol. 2017;28:xii44–55. doi:10.1093/annonc/mdx237.
- Choi Y-E, Yu H-N, Yoon C-H, Bae Y-S. Tumor-mediated down-regulation of MHC class II in DC development is attributable to the epigenetic control of the CIITA type I promoter. Eur J Immunol. 2009;39(3):858–868. doi:10.1002/eji.200838674.