
ABSTRACT
Intestinal epithelial barrier protects intestine from infection and injury, while chronic inflammation is a trigger for tumorigenesis. As a member of tight junctions (TJs) family, Claudin-7 (Cldn-7) is dedicated to maintaining cell polarity and TJs barrier integrity, and closely related to the development of inflammation and tumors. However, potential roles of Cldn-7 in intestinal inflammation and colitis-associated colorectal cancer (CAC) have not been well characterized in vivo. Here, we analyzed the expression profile of Cldn-7 in inflammatory bowel disease (IBD) and CAC. Colitis and colitis-cancer transformation models were established based on inducible intestinal conditional Cldn-7 gene knockout mice (Cldn7fl/fl;villin-CreERT2), by intraperitoneal injection of azomethane (AOM) and dextran sodium sulfate (DSS) feeding. Cldn-7 knockout promoted susceptibility to colitis and CAC, aggravated clinical symptoms, severely damaged intestinal epithelium, increased mucosal inflammation accompanied dysregulated cell proliferation-apoptosis. Epithelial barrier integrity was destroyed, and intercellular permeability was increased. After AOM/DSS induction, tumor burden and volume were increased, characterized by enhanced proliferation and activation of Wnt/β-catenin signaling pathway. Mechanistically, Cldn-7 deficiency promoted colitis and subsequently malignant transformation by destroying TJs integrity and increasing inflammatory cascade. Overall, based on Cldn-7 knockout mouse model, we have first demonstrated the key roles of Cldn-7 in maintaining intestinal homeostasis and preventing IBD and consequent CAC.
Abbreviations: AJs: adherens junctions; AOM: azomethane; Cldn-7: Claudin-7; CRC: colorectal cancer; CAC: colitis-associated colorectal cancer; CD: Crohn’s disease; DSS: dextran sodium sulfate; DAI: disease activity index; EMT: epithelial-mesenchymal transition; FITC: fluorescence isothiocyanate; HB: hemoglobin; IBD: inflammatory bowel disease; IECs: intestinal epithelial cells; ISCs: intestinal stem cells; PLT: platelet; RBC: red blood cell; ROS: reactive oxygen species; TAM: tamoxifen; TJs: tight junctions; TCF/LEF: T-cell factor/lymphoid enhancer factor; UC: ulcerative colitis; WBC: white blood cell.
Introduction
Inflammatory bowel disease (IBD), which comprises Crohn’s disease (CD) and ulcerative colitis (UC), has a steadily increasing incidence in newly industrialized countries.Citation1 It is a group of chronic recurrent intestinal inflammatory lesions caused by nonspecific pathogens, and its pathogenesis involves abnormal immune response to intestinal microbiota due to genetic susceptibility. Intestinal mucosal immunity, genetics, microorganisms, and uncertain environmental factors disrupt intestinal homeostasis leading to intestinal inflammation.Citation2,Citation3 Chronic inflammation is a major cause of tumor formation, and IBD is a primary driving force for progression of colorectal cancer (CRC). Biologically, IBD results in anti-tumor immune dysfunction, colonic mucosa destruction, cell proliferation and malignant transformation.Citation4,Citation5 Particularly, duration and severity of intestinal inflammation are positively correlated with increased risk of colitis-associated colorectal cancer (CAC).Citation6
Intestinal barrier function as a hallmark of health. Intestinal barrier dysfunction, increased permeability of epithelial cells and invasion of intestinal lumen antigens are related to intestinal inflammatory imbalance and tumorigenesis.Citation7,Citation8 To maintain intestinal homeostasis, intestinal epithelial cells (IECs) form an intestinal barrier which consists special structures of tight junctions (TJs) and adherens junctions (AJs), which acts as a defense system against pathogens and toxic substances. These proteins provide mechanical junctions between neighboring cells, allowing bi-directional transport of ions, liquids and nutrients, as well as preventing bacteria from activating mucosal immune system.Citation9
As an important part of intestinal epithelial barrier, TJs are composed of multiple transmembrane proteins. TJ-related proteins affect intestinal permeability, the development of intestinal inflammation and CAC.Citation10–17 Claudins maintain the integrity of intestinal barrier and epithelial cell polarity, and regulate permeability.Citation18 Additionally, claudins may also act as signaling proteins involved in inflammation, cell proliferation and differentiation.Citation19 Tight junction protein claudin-7 (Cldn-7), a member of claudins family, is associated with intestinal inflammation and tumors. Cldn-7 is down-regulated in intestinal inflammatory disorders.Citation20 Cldn-7 knockout resulted in an imbalance in the paracellular flux of intestinal small organic solute, promoted degradation of intestinal extracellular matrix and enhanced mucosal immunity.Citation13,Citation14 Our previous study confirmed that Cldn-7 is a potential tumor suppressor. The worse the tumor differentiation, the more obvious the down-regulation of Cldn-7, and the decrease of Cldn-7 may indirectly regulate CRC cell adhesion, induce tumor invasion and metastasis by promoting epithelial-mesenchymal transition (EMT).Citation21 Controversially, Cldn-7 is reported to be pro-oncogenic by inhibiting cell apoptosis and enhancing tumor aggressiveness.Citation22,Citation23 Therefore, potential roles of Cldn-7 in mucosal inflammation and tumor progression remain to be clarified.
In this study, we explored the role and molecular mechanisms of Cldn-7 in IBD and CAC based on a pre-constructed gene knockout mouse model. Notably, Cldn-7 knockout enhanced sensitivity to dextran sodium sulfate (DSS)-induced colitis and azomethane (AOM)/DSS-induced CAC. For the first time, we have demonstrated that Cldn-7 plays a critical role in maintaining the integrity of intestinal epithelial barrier and intestinal homeostasis to inhibit the progression of colitis and CAC.
Results
Cldn-7 expression is decreased in inflamed mucosa of IBD patients and DSS-induced colitis mouse model
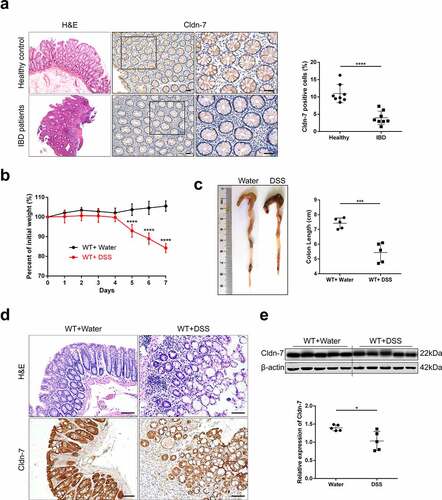
To examine expression profiling of Cldn-7 in inflammatory mucosa of IBD patients, colonic biopsies of IBD patients and healthy volunteers were randomly selected. As shown in , compared with healthy control group, intestinal mucosa of IBD patients was destroyed, presented with massive inflammatory cell infiltration in lamina propria and formation of crypt abscesses. Immunohistochemistry staining confirmed that the number of Cldn-7 positive cells in inflammatory mucosa was significantly reduced (P < .0001). For the in vivo study, C57BL/6 wild-type mice were treated with 5%DSS in drinking water for 7 days to induce colitis. Compared with control mice, DSS-fed mice lost more body weight and had shorter colon length (). In addition, intestinal inflammation, mucosal destruction, vacuole formation and inflammatory cell infiltration in DSS-fed mice were more obvious. Cldn-7 expression of colonic epithelium in DSS-treated mice was slightly down-regulated (P < .05, ), consistent with that in IBD patients.
Figure 1. Expression profile of Cldn-7 in inflamed mucosa of IBD patients and DSS-induced colitis mouse model. (a) Representative intestinal sections were prepared from colonic tissues of healthy control and inflamed mucosa of IBD patients, and stained for Cldn-7 by immunohistochemistry. Right: Quantification of the average intensity of Cldn-7 per high power field (HPF) (****p < .0001, n = 8). (b) 5%DSS was administered in drinking water for 7 days. Changes in body weights were shown as percentage of initial weights at the start of the experiments (****p < .0001, n = 5). (c) Gross morphology and length (from appendix to anus) of colon tissues at the end of the experiment (***p < .001, n = 5). (d) Representative images of H&E-stained colonic tissues and expression of Cldn-7 by IHC (n = 5). (e) Representative immunoblot images of Cldn-7 from colonic lysates at the end of DSS treatment (upper panel). Lower panel: quantitative expression of Cldn-7 (*p < .05, n = 5). All images were visualized using Nikon Eclipse E200 microscope and analyzed by Image J software. Scale bars: 50 µm
Intestinal epithelial-specific knockout of Cldn-7 increases susceptibility to DSS-induced colitis
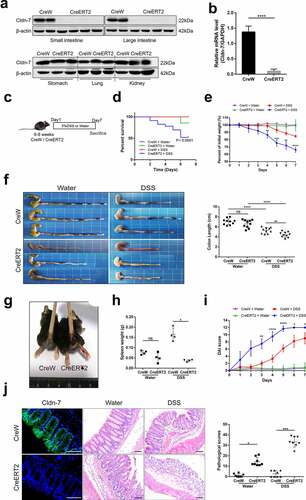
To further determine whether down-regulation of Cldn-7 promotes intestinal inflammation, inducible intestinal conditioned Cldn-7 knockout (Cldn7fl/fl;villin-CreERT2, abbreviated as CreERT2) and control (Cldn7fl/fl;villin-CreW, abbreviated as CreW) murine models were constructed, tamoxifen (TAM) was administered by intraperitoneal injection (1 mg in 100ul sterile sunflower oil, every 4 days). The knockout efficiency of Cldn-7 was verified after 10 consecutive inductions. The results showed that Cldn-7 expression was absent in intestinal epithelium (but intact in stomach, lung and kidney) after tamoxifen induction, as schematically presented in . At the same time, the mRNA level of Cldn-7 was remarkably reduced in colonic tissue (P < .0001, ), verifying the Cldn-7 knockout was intestinal-specific. CreERT2 mice developed more serious intestinal diseases, presenting with a lower survival rate, more serious body weight loss, colon shortening, rectal bleeding, spleen weight loss, and increased disease activity index (DAI) scores than CreW mice after DSS exposure (). Histological analysis and colonic pathology score also revealed significantly higher levels of intestinal inflammation in CreERT2 mice, accompanied by epithelial mucosal destruction, glandular crypt structure damage, and inflammatory infiltration. Due to the lower concentration of DSS, the pathological changes of CreW mice were slight (). Taken together, these data indicated that deletion of Cldn-7 might aggravate intestinal damage induced by DSS.
Figure 2. Increased susceptibility of Cldn-7 knockout mice to DSS-induced colitis. (a) Western blot experiments were performed in tissues from CreERT2 and CreW mice. (b) Relative mRNA expression level of Cldn-7 in colonic epithelia (****p < .0001). (c) Schematic protocol used to induce colitis. CreW and CreERT2 mice were fed 3%DSS or tap water for 7 days. (d) Kaplan-Meier survival plots described colitis-induced animal mortality (CreERT2+DSS vs CreW+DSS, p < .0001). (e) Changes in body weights were measured as percentage of the initial weight at the start of the experiments (CreERT2+DSS vs CreW+DSS, **p < .01, ***p < .001, ****p < .0001, n = 6). (f) Gross morphology and length of colon from mice either untreated or treated with DSS for 7 days (**p < .01, ****p < .0001, n = 10). (g) Representative photograph of rectal bleeding from the mice treated with DSS. (h) Spleen weights (*p < .05, n = 4). (i) Daily recorded DAI score (CreERT2+DSS vs CreW+DSS, *p < .05, **p < .01, ****p < .0001, n = 6). (j) Representative H&E-stained colon sections and colonic pathological scores for inflammation, extent of injury, crypt damage and regeneration (*p < .05, ***p < .001, n = 8). Images were acquired with Nikon Eclipse 80i microscope. Scale bars: 50 µm
Loss of Cldn-7 increases colonic infiltration of leukocytes during experimental colitis
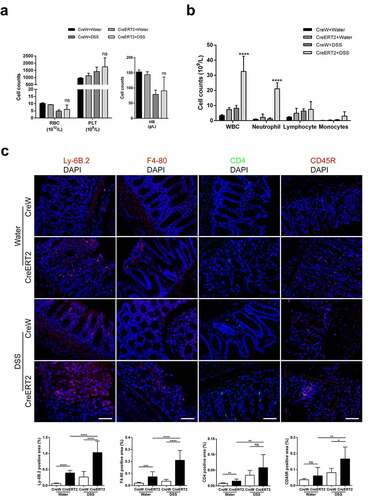
Next, we investigated whether disease progression and obvious mucosal injury in DSS-treated CreERT2 mice were caused by intensified mucosal inflammation. Before we sacrificed the mice, routine blood test was performed. Red blood cell (RBC) count and hemoglobin (HB) count were reduced whereas platelet (PLT) count was increased due to increased bloody stools in DSS group, but the difference was not significant (). Furthermore, the number of white blood cell (WBC) and neutrophils was conspicuously higher in CreERT2+DSS mice (). Next, we observed that the proportion of Ly-6B.2-positive neutrophils and F4-80-positive macrophages were increased significantly in colonic mucosal layer of CreERT2+DSS mice as compared to CreW+DSS mice, while CD4-positive T cells and CD45R-positive B cells only had an upward trend as detected by immunofluorescence ().
Figure 3. Loss of Cldn-7 increases colonic infiltration of leukocytes during experimental colitis. (a) Changes of hematological parameters in mice after induction of experimental colitis (n = 3–4). (b) Quantification of leukocytes (CreERT2+DSS vs CreW+DSS, ****p < .0001, n = 3–4). (c) Neutrophils, Macrophages, T cells and B cells were visualized by immunolabeling specific cellular markers, Ly-6B.2 (red), F4-80 (red), CD4 (green) and CD45R (red), correspondingly. DAPI was used to visualize nucleus (blue). Quantification of positive area was shown in the lower panels (n = 3). Scale bars: 25 µm. The symbols *, **, *** and **** indicate p < .05, p < .01, p < .001 and p < .0001, respectively. Images were acquired with Nikon Eclipse 80i microscope and analyzed by Image J software
Cldn-7 deficiency dysregulates cytokine and chemokine expression following DSS treatment
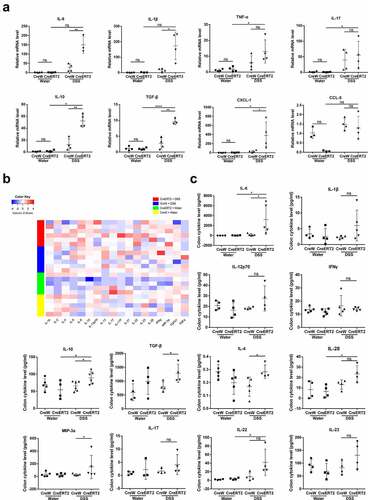
In addition, expression profile of inflammatory cytokines in colonic epithelia was detected by qRT-PCR. After DSS treatment, IL-6, IL-1β, IL-10, TGF-β and chemokine CXCL-1 in colonic mucosa of CreERT2 mice were up-regulated compared with CreW group (P < .05, ). Based on Array analysis, IL-6, IL-10, TGF-β, IL-4 and MIP-3a were increased in CreERT2 mice after DSS exposure (P < .05, ). Together, these results provided further evidence of high-scale mucosal inflammation in CreERT2 mice, and revealed that Cldn-7 deficiency promoted intestinal inflammatory immune response in DSS-induced colitis.
Figure 4. Cldn-7 deficiency dysregulates cytokine and chemokine expression. (a) Total RNA was extracted from colonic tissues, relative mRNA expression levels of inflammatory cytokines in colonic epithelia were determined by qRT-PCR, including IL-6, IL-1β, TNF-α, IL-17, IL-10, TGF-β, and chemokine CXCL-1, CCL-5 (*p < .05, **p < .01, ****p < .0001, n = 3–4). (b) Heat map indicating the cytokines expression profiles. Each row represents expression profile of a tissue sample, and each column corresponds to a change in protein level. The color scale from blue to red indicates z score. (c) Cytokines were quantified with Quantibody® Mouse TH17 Cytokine Array 1. The results were analyzed using RayBio Analysis Tool Excel sheet (*p < .05, n = 4–6)
Deletion of Cldn-7 affects intestinal homeostasis and disrupts intestinal barrier function
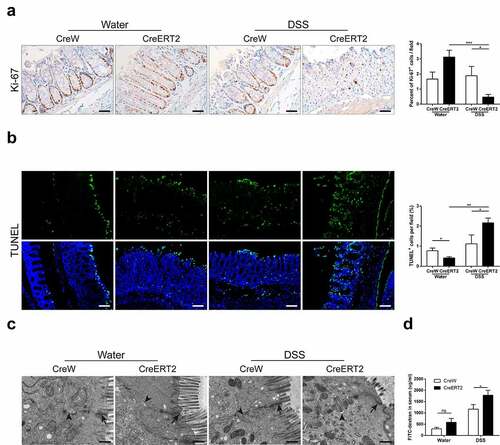
Given the increased intestinal inflammation in CreERT2 mice, potential roles of Cldn-7 in regulating intestinal homeostasis and mucosal permeability were determined. Ki-67 and TUNEL staining were used to detect proliferation and apoptosis of colonic epithelium. As shown in , Ki-67-positive epithelial cells in CreW mice were normally limited to crypt region. However, in CreERT2 mice with normal drinking, cell proliferation was extended from crypt to lumen along crypt-surface axis. After DSS exposure, intestinal tract of CreERT2 mice was severely damaged and proliferation was significantly reduced (P < .05). Apoptotic intestinal epithelial cells generally occurred in villi on the surface of intestinal epithelium and fell off into intestinal lumen. Apoptosis was increased significantly and involved crypt area in CreERT2+DSS group (P < .05, ). These findings verified increased intestinal damage and delayed recovery of colonic epithelium in Cldn-7 knockout mice. Electron microscopy identified intact TJs, dense and neatly arranged intestinal villi, and slightly widened intercellular space in CreERT2+Water group. After DSS treatment, TJs were damaged, whereas villi were sparse and fractured, and intercellular gap obviously widened (). The serum fluorescence isothiocyanate (FITC)-dextran concentration (µg/ml) representing intestinal permeability was markedly increased in CreERT2 mice after DSS exposure (P < .05, ). Thus, Cldn-7 deletion might destroy TJs integrity and increase mucosal permeability, leading to epithelial barrier dysfunction.
Figure 5. Deletion of Cldn-7 affects intestinal homeostasis and exacerbates intestinal barrier function. (a) Epithelial cell proliferation was assessed by immunostaining of Ki-67. Right: Ki-67-positive cells were quantified in at least five HPFs per mouse. Scale bars: 25 µm (*p < .05, ***p < .001, n = 3). (b) The number of apoptotic cells was visualized using TUNEL assay (green). Nuclear counter-staining (blue) was used to visualize the position of an individual cell. Scale bars: 50 µm. Right: Percentage of apoptotic cells per HPF was averaged in each group (*p < .05, **p < .01, n = 4). Images were acquired with Nikon Eclipse 80i fluorescence microscope and analyzed by Image J software. (c) Representative electron microscope images identified tight junctions and intercellular spaces of colonic epithelium. Black arrows label tight junctions and arrowheads delineate intercellular spaces on the basolateral side. Scale bars: 500 nm. (d) Intestinal permeability was determined by measuring transmucosal flux of FITC-dextran in blood serum after four hours of gavage with FITC-dextran (*p < .05, n = 4)
Cldn-7 deficiency exacerbates tumorigenesis in the AOM/DSS-induced colitis-associated colorectal model
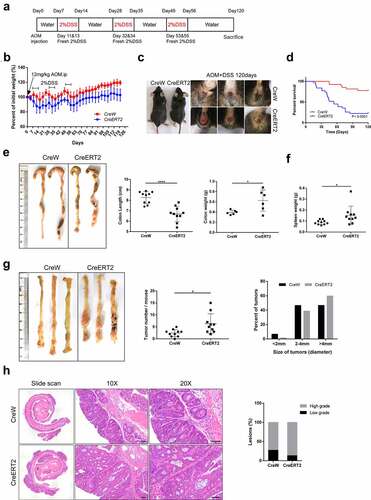
We have reported that Cldn-7 was down-regulated in CRC tissue. To investigate whether Cldn-7 loss could contribute to intestinal tumorigenesis, AOM/DSS induction was used to construct a murine model of CAC (). During CAC induction, Cldn-7 knockout mice displayed significantly enhanced weight loss, worsened rectal prolapse, decreased survival rate, shortened colon length, as well as increased colon tumor burden (P < .05), and the tumor size was larger in the CreERT2 group (CreERT2 vs CreW = 59.7% vs 46.7% in the >4 mm group, ). Histological analysis revealed that Cldn-7 deletion caused a higher proportion of high-grade tumors compared with CreW littermates (86.6% vs 72.7%), but the difference was not statistically significant (). These results suggested that Cldn-7 deficiency was associated with inflammation-associated tumorigenesis.
Figure 6. Susceptibility to carcinogenesis is enhanced in Cldn-7-deficient mice in the AOM/DSS-induced colitis-associated colorectal model. (a) Schematic representation of AOM/DSS treatment to induce CAC. (b) Changes of body weight curve in AOM/DSS induction period. (c) Representative images of gross morphology and anal fistula at the end point of 120 days. (d) Survival analysis at the end of the experiment. (e) Representative views of colons, lengths and weights of colons at day 120 after AOM injection (*p < .05, ****p < .0001, n = 6–10). (f) Spleen weights (*p < .05, n = 10). (g) Representative images of colon tumors in AOM/DSS-induced mice. The number of lesions in the entire colon per mouse was counted, and the size of each lesion was measured (*p < .05, n = 10). (h) Images of H&E-stained colon sections demonstrated histologic features of tumors in CreW and CreERT2 mice (Scale bars: 50 µm). Right: Tumors were classified into low or high grade based on histological analysis. Images were visualized using Nikon Eclipse E200 microscope
Loss of Cldn-7 activates Wnt/β-catenin pathway in colonic tissues of AOM/DSS mice
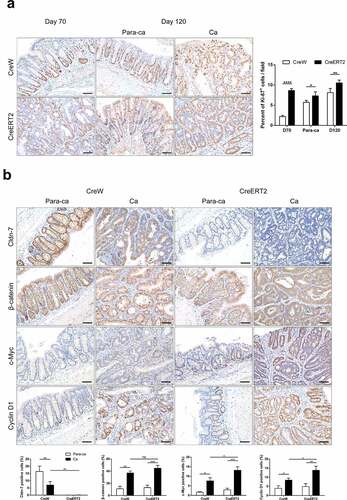
To explore molecular mechanisms underlying how Cldn-7 participate in intestinal carcinogenesis, potential effects of Cldn-7 deletion on cell proliferation and Wnt/β-catenin pathway were evaluated. Consistent with colon tumor burden, epithelial proliferation rate in tumor tissues and paracancerous tissues derived from CreERT2 group was higher than that of control group (P < .05, ). It is well known that β-catenin dysregulation is a key indicator of Wnt signaling overactivation. In paracancerous tissues, β-catenin was expressed on the cell membrane, and nuclear c-Myc and Cyclin D1 were restricted to cells at the base of crypts. The expression of β-catenin and downstream target genes c-Myc and Cyclin D1 were significantly up-regulated in cancer tissues as compared to adjacent tissues, and β-catenin showed cytoplasmic and nuclear localization (P < .05, ). Cldn-7 deletion further promoted the expression of c-Myc and Cyclin D1, while β-catenin only had an upward trend compared with CreW tumor tissues, and there was no statistical difference. It suggested that Cldn-7 knockout might promote proliferation of intestinal epithelial cells and activate Wnt/β-catenin signaling pathway to spur CAC development.
Figure 7. Loss of Cldn-7 activates Wnt/β-catenin signaling pathway. (a) Paracancerous tissues and tumor sections were stained for Ki-67 on day 70 and 120, respectively, during experimental procedure. Right: Quantification of positive Ki-67 area per field (*p < .05, **p < .01, ****p < .0001, n = 5). (b) Representative immunohistochemical images of Cldn-7, β-catenin, c-Myc and Cyclin D1 in cancer tissues and adjacent noncancerous tissues from CreW and CreERT2 mice at the end point of 120 days. Lower panels: Percentage of positive cells was analyzed using ImageJ software (*p < .05, **p < .01, ***p < .001, n = 3). Scale bars: 50 µm. All images were visualized using Nikon Eclipse E200 microscope and analyzed by Image J software
Knockdown of Cldn-7 disrupts biological function and aggravates inflammatory injury in DSS induced cytotoxicity
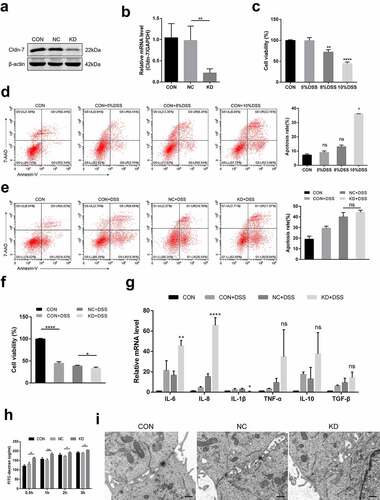
To investigate biologic functions of Cldn-7 in DSS colitis at the cellular level in vitro, Caco-2 cells were transfected with lentivirus to knockdown Cldn-7. Western blot and qRT-PCR were performed to verify the efficiency of knockdown (). Injury was induced by different concentrations of DSS (5%, 8%, and 10%, respectively). Based on CCK-8 and flow cytometry, cell viability was significantly decreased whereas apoptosis increased with elevated DSS concentration (). Since 10%DSS caused a remarkable decrease in cell activity, it was selected for subsequent induction of inflammatory damage to Caco-2 cells. Cldn-7 gene knockdown reduced cell viability (P < .05), and slightly increased the cell apoptosis, but the difference was not significative (). Meanwhile, qRT-PCR results demonstrated that Cldn-7 knockdown increased the production of IL-6 and IL-8 after DSS treatment (P < .01), the expression of IL-10 and TNF-α was up-regulated, but there was no statistical difference (). The above findings indicated that down-regulation of Cldn-7 increased DSS-induced inflammatory injury. Based on permeability assay, FITC flux was increased after Cldn-7 knockdown, while fuzzy TJs structure was displayed on electron microscope (). Therefore, Cldn-7 down-regulation might destroy intestinal epithelial barrier and stimulate inflammatory damage in vitro.
Figure 8. Inhibition of Cldn-7 disrupts biological function and aggravates inflammatory injury in DSS induced cytotoxicity. (a-b) Transfection of Caco-2 cells with specific shRNA. Efficiency of knockdown was confirmed by WB and qRT-PCR (**p < .01). (c) The effect of DSS at different concentrations (5%, 8%, 10%, respectively) on cell viability was estimated by CCK-8 assay (**p < .01, ****p < .0001). (d) Potential effect of DSS on cell apoptosis was determined by Annexin V-PE/7-AAD staining assay (left panels). Quantification of the number of apoptotic cells was performed (right panel, * p < .05). (e) The effect of Cldn-7 knockdown on cell apoptosis. (f) The effect of Cldn-7 knockdown on DSS-induced cell viability (*p < .05, ****p < .0001). (g) The mRNA levels of cytokines in DSS-stimulated injury of epithelial cells were analyzed by qRT-PCR (*p < .05, **p < .01, ****p < .0001). (h) Effects of Cldn-7 on intestinal permeability in Caco-2 cells. After cells grew into a monolayer, FITC-dextran was added to the upper chamber, and individual concentration of FITC-dextran in the lower chamber was detected at 0.5 h, 1 h, 2 h and 3 h, respectively (*p < .05, **p < .01). (i) Representative electron microscope images described potential effects of Cldn-7 on tight junctions and intercellular spaces of Caco-2 cells. Scale bars: 5 μm. For all experiments, data are representative of at least three independent experiments
Discussion
TJs constitute an intercellular adhesion complex that plays a crucial role in maintaining gastrointestinal mucosal barrier and intestinal homeostasis. TJs maintain cell polarity as well as regulate paracellular permeability and water flux by limiting diffusion of lipids and proteins within plasma membrane.Citation9 Epithelial barrier dysfunction is a vital pathogenic factor in gastrointestinal diseases including IBD and CRC. Thus, destruction of intestinal barrier leads to activation of specific signaling pathways, which can initiate intestinal inflammation and tumor formation.Citation24,Citation25 Changes in the integrity of TJs and increases in intestinal mucosal permeability precede and exacerbate intestinal inflammation, allowing microbiota invasion and stimulating immune cells to produce inflammatory antigens.Citation26 Notably, continuous extensive colitis is a crucial cause of malignant transformation in colon and rectum.Citation27
Cldn-7 is abnormally expressed in intestinal inflammation and CRC, and is involved in cell proliferation and malignant transformation. Our previous research found that global Cldn-7 knockout mice had damaged intestinal epithelial integrity, aggravated intestinal inflammation, and upregulated the expression of matrix metalloproteinases.Citation13 Furthermore, intestinal conditional Cldn-7 knockout mice spontaneously developed atypical hyperplasia and adenomas.Citation28 Cldn-7 expression in CRC tissue was closely related to the grade of tumor differentiation. In addition, down-regulation of Cldn-7 promoted xenograft tumor growth, weakened cell-cell adhesion, and promoted invasion and metastasis of CRC.Citation21 Cldn-7 may be used as a prognostic marker for tumor development and disease-free survival.Citation29,Citation30 Tanaka et al. revealed that Cldn-7 knockout increased intestinal cell apoptosis and neutrophil infiltration, as well as elevated paracellular flux of small organic solvents.Citation14 Cldn-7 expression was restored and intestinal epithelial recovery was facilitated when drugs applied to alleviate intestinal injury.Citation31 These studies prompted us to explore potential roles of Cldn-7 in maintaining intestinal homeostasis and tumorigenesis in vivo.
Animal models can reshape human diseases and have great significance in exploring molecular mechanisms underlying various diseases. DSS-induced colitis can mimic IBD in histopathology, the chemical inflammatory agent DSS has direct toxicity to intestinal epithelial cells, induces mucosal damage and affects mucosal barrier integrity.Citation32 In this study, Cldn-7 expression in colonic epithelial cells of IBD patients was significantly down-regulated compared with healthy individuals, consistent with that in a murine model of colitis. Deletion of IEC-specific Cldn-7 aggravated DSS-induced colon inflammation, resulting in reduced tolerance to DSS, severe intestinal epithelial injury and basal crypt destruction, as well as intensified immune cell infiltration. Routine blood examination confirmed specific phenotype of CreERT2+DSS mice with more serious inflammation response. In addition, up-regulation of chemokine CXCL-1 can drive recruitment of neutrophils to inflammation sites. Our previous studies have found that the expression of Cldn-4 was decreased, Cldn-15 and occludin were up-regulated, while Cldn-1, Cldn-3, and E-cadherin showed no significant changes in Cldn-7 knockout mice, which further verified the function of Cldn-7 in maintaining intestinal barrier.Citation33 Thus, destructed TJs, increased intestinal permeability, and up-regulated inflammatory cytokines jointly induced inflammatory cascade, which exacerbated damage to colon tissue as well as deterioration of pathology. Furthermore, increased release of inflammatory cytokines, together with intestinal epithelial cell proliferation/apoptosis imbalance feedback intensified the destruction of TJs, leading to loss of intestinal barrier. This TJs-inflammatory cycle could be observed in IBD patients.Citation34
AOM is a chemical carcinogen, AOM/DSS model simulates the pathophysiological process of CAC.Citation35 AOM induced β-catenin mutation and promoted β-catenin translocation to the nucleus, which interacts with T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factor family, leading to abnormal activation of Wnt signaling pathway and downstream target genes c-Myc and Cyclin D1.Citation36 The Wnt/β-catenin signaling pathway plays a crucial role in coordinating cell proliferation, stem cell differentiation and cell migration.Citation37 Consistent with down-regulation of Cldn-7 in tumor tissue, Cldn-7 deletion promoted disease progression of AOM/DSS-induced CAC. Compared with CreW littermates, CRC tumor burden, high-grade intraepithelial neoplasia, tumor cell proliferation as well as expression of Cyclin D1 and c-Myc were significantly enhanced in Cldn-7-deficient mice. Inflammation is an important part of tumor microenvironment in CRC, both damage and repair of colon epithelium will mediate a mismatch of genome replication. The enrichment of immune cells can produce a large amount of reactive oxygen species (ROS) to promote DNA damage. Pro-oncogenic cytokines can activate carcinogenic signaling pathways and aggravate malignant proliferation and carcinogenesis.Citation38,Citation39 Deletion of Cldn-7 promoted the progression of intestinal inflammation, abnormal cell proliferation and tumor occurrence partially by activating Wnt/β-catenin signaling pathway.
A key question that needs to be further explored is whether increased colitis severity and tumor formation in Cldn-7-deficient mice are associated with delayed intestinal remodeling. Intestinal tissue remodeling depends on the ability of intestinal stem cells (ISCs) to differentiate into normal epithelial cell lineages. Cellular damage and repair disorders increase the risk of carcinogenesis.Citation40 We have found that Cldn-7 has a regulatory role in small intestinal stem cells, which controls the survival, self-renewal and epithelial differentiation of ISCs.Citation33 We speculate persistent colon inflammation and dysplastic intestinal epithelium may correlate with inability of stem cells to differentiate in Cldn-7 knockout mice. Colonic stem cells are responsible for mucosal regeneration and the maintenance of stable colonic luminal environment, while the inner mucosa in turn protects colonic stem cells from damage by luminal contents.Citation41 Cldn-7 contributes to the maintenance of colonic epithelial integrity, whether there is a certain regulatory effect on colonic stem cells is unknown. In the next stage, we will explore the molecular mechanisms of how Cldn-7 and ISCs microenvironment influence the development and progression of CRC.
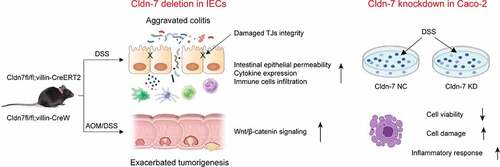
In summary, our findings reveal inhibitory effects of Cldn-7 on occurrence of IBD and CAC, Cldn-7 is an essential regulator of intestinal homeostasis. Loss of Cldn-7 destroys intestinal epithelial barrier, alters intestinal epithelial injury/repair homeostasis, and promotes inflammatory signaling cascade, leading to abnormal proliferation and tumorous transformation ().
Figure 9. Working model of the role of Cldn-7 in regulating intestinal homeostasis and colitis-associated carcinogenesis
Materials and methods
Human samples
The colonoscopy biopsies of IBD patients and healthy people were obtained from pathology database of Beijing Shijitan Hospital. The study has been performed in accordance with the Declaration of Helsinki and approved by the Clinical Research Ethics Committee of Capital Medical University Affiliated Beijing Shijitan Hospital (2018KY(55)). All subjects have provided written informed consent.
Animals
Six to eight-week-old male wild-type C57BL/6 mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. Inducible intestinal conditioned Cldn-7 gene knockout mice (CreERT2) were generated using Cre/Loxp system by our team as reported previously.Citation28 All mice were maintained under specific pathogen-free conditions in the animal facilities of Beijing Shijitan Hospital. All mice were given a standard chow diet and tap water ad libitum, and randomized into groups for subsequent in vivo experiments at 8–10 weeks. All experiments followed the ARRIVE guidelines and approved by Animal Ethics Committee of Beijing Shijitan Hospital Institutional Review Board (2018KY(55)).
Induction and characteristics of DSS-colitis
The DSS-induced colitis mouse model was established as previously described.Citation42 Briefly, C57BL/6 (WT) mice were treated with 5%DSS (molecular weight, 36–50 kDa, MP Biomedicals, LLC; Solon, OH, USA) dissolved in drinking water. The CreERT2 and littermate CreW mice were fed with 3%DSS for 7 days. Unchallenged animals received tap water. DAI score is scored based on three parameters: body weight loss, stool consistency and intestinal bleeding.Citation43 Briefly, body weight loss scores were determined as follows: no weight loss was scored as 0, 1%-5% weight loss as 1, loss of 5%-10% as 2, 10%-15% loss as 3, more than 15% was scored as 4. For stool scores, well-formed pellets were scored as 0, semi-formed stools as 2, watery diarrhea as 4. For intestinal bleeding, no bleeding as 0, slight bleeding and positive hemoccult as 2, gross bleeding was scored as 4. All mice were euthanized according to criteria of IACUC, when colon and rectum were removed, colon length was measured, and the colonic tissues were separated into several segments for paraffin embedding or cryopreservation.
AOM/DSS-induced CAC carcinogenesis model
AOM/DSS-induced colorectal cancer was established as described previously.Citation44 At 8–10 weeks of age, CreERT2 and CreW mice were intraperitoneally injected with TAM (T5648, Sigma-Aldrich, St. Louis, MO) to induce recombination, and then injected with 12 mg/kg carcinogen AOM (A5486, Sigma-Aldrich, St. Louis, MO). After 7 days of AOM intervention, the mice were administered with 2%DSS continuously for 7 days, followed by regular water for 2 weeks. This cycle of DSS was repeated three times. Once the mice began to deteriorate, lost more than 20% of the baseline body weight, or showed hunched posture, weakness or inability to move, they were euthanized. During the study period, mice were monitored for body weight, fecal bleeding, rectal prolapse and survival status. At 120 days, all mice were sacrificed, spleen weight, colon weight and colon length were measured. The colon was opened longitudinally to quantify tumor nodules and measure the size of tumor. The colonic tissues were fixed in 10% formalin for histopathological analysis. The sections were stained with hematoxylin and eosin (H&E) for tumor characteristics evaluation by gastrointestinal pathologists blinded to treatment.
Western blot analysis (WB)
Proteins were extracted from intestinal tissues using ice-cold RIPA lysis buffer supplemented with protease inhibitors. Total protein concentration was quantified by BCA assay (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of proteins were denatured for 5 min and separated by 12% SDS-PAGE, and then electrotransferred onto nitrocellulose (NC) membrane (Millipore) using standard procedures. Afterward, NC membrane was blocked with 5% skim milk powder in TBST, then incubated with primary Cldn-7 antibody and corresponding secondary antibody. Finally, quantification of WB band intensity was conducted using the Odyssey CLxInfrared Imaging System (LI-COR, USA). The corresponding antibody information is listed in .
Table 1. Antibody information used in this experiment
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated from colonic segments of mice or Caco-2 cells using TRIzol reagent. The RNA (1 μg) was reversed to cDNA using qPCR RT Master Mix (TOYOBO, Japan) according to the manufacturer’s protocol. Then, qRT-PCR was performed using SYBR Green Master Mix (Applied Biosystems, USA). The primer sequences designed by Sangon Biotech are listed in . Relative expression of each gene was calculated by comparative threshold cycle (2−ΔΔCt) method and normalized to β-actin (internal control).
Table 2. Primer sequences used for the qRT-PCR assays
Histopathological analysis
For histological assessment, the mice were sacrificed and colonic tissues were removed, longitudinally opened, rolled into a “swiss roll”, fixed immediately overnight, and subsequently embedded in paraffin. The colonic tissue specimens were stained with H&E, and intestinal tissue lesions were analyzed under a microscope. Histopathology scores were recorded by pathologists blinded to experimental design according to previous criteria based on four parameters (inflammation, extent, crypt damage and regeneration).Citation45
Immunohistochemistry (IHC) and immunofluorescence (IF)
Immunohistochemistry was performed to determine expression profile of proliferation biomarker (Ki-67) and Wnt signaling (β-catenin, c-Myc, Cyclin D1). Biomarkers for infiltrated neutrophil (Ly-6B.2), macrophage (F4-80), T lymphocyte (CD4) and B lymphocyte (CD45R) were evaluated by immunofluorescence. Tissue slides underwent dewaxing, rehydration, antigen retrieval, and incubated with 3% hydrogen peroxide (H2O2) to inactivate endogenous peroxidase activity. Tissue sections were blocked with 3% bovine serum albumin (BSA) for 1 h at RT, and incubated with primary antibodies and secondary antibodies. For IF, cell nucleus was counterstained with DAPI. For IHC, DAB was used as the final chromogen and hematoxylin was used to counterstain nuclei. All images were collected under a microscope.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
Apoptosis of intestinal epithelial cells was evaluated using One Step TUNEL Apoptosis Assay Kit (Beyotime Biotechnology, Shanghai, China) in accordance with the manufacturer’s instructions. Apoptotic cells were observed by a fluorescence microscope, Image J software was used to record the percentage of positive cells for image analysis and quantification.
Determination of permeability in vivo and in vitro
In vivo epithelial permeability was assessed by FITC-dextran (4 kDa, 46944, Sigma-Aldrich, St.Louis, MO) as previously described.Citation46 Mice were treated with fasting for solids and liquids for 4 h before gavage; then, mice were gavage with FITC-dextran (60mg/100g body weight) and euthanized after 4 h to collect blood via cardiac puncture. Serum was obtained by centrifugation. For in vitro permeability assay, Cldn-7 knockdown Caco-2 cells and negative control cells in the logarithmic growth phase were seeded onto permeable polycarbonate inserts in 24-well Transwell® plates (pore size; 0.4 µm, growth area; 0.33 cm2, Corning, Inc., Corning, NY, USA) at a density of 2 × 104 cells/well.Citation47 The cells were maintained in an incubator for 3 weeks to form a confluent monolayer. 100 μl of 1 mg/mL FITC-dextran was added to the upper chamber, the basolateral chamber was filled with 600 μl of PBS. Then, 100 μl basolateral medium was collected and transferred to a 96-well plate at 0.5, 1, 2 and 3 h, respectively. Simultaneously, an equal volume of fresh PBS was added to the basolateral compartment. Fluorescence intensity was quantified using fluorescence microplate reader (Tecan Group Ltd, Männedorf, Switzerland) at 485 nm excitation and 535 nm emission wavelengths. The concentration of FITC-dextran was calculated according to standard curve generated by a serial dilution of FITC-dextran in PBS.
Electron microscopy analysis
Fresh intestinal tissues and confluent Caco-2 cells were fixed in 2.5% glutaraldehyde solution at 4°C for 4 h, and rinsed with 0.1 M phosphate buffer (pH7.4) three times. The samples were post-fixed with 1% osmium tetroxide, dehydrated by graded ethanol and acetone series. Permeation was carried out with a mixture of acetone and embedding agent (1:1, 2:1 and 0:1 proportion). The samples were transferred to pure embedding agent and infiltrated overnight in a 37°C oven and polymerized at 60°C for 48 h in an incubator. Ultrathin sections with a thickness of 70 nm were prepared by EM UC7 ultramicrotome (Leica, Wetzlar, Germany) and double stained with 2% uranyl acetate and lead citrate solution. Images were observed and captured under a HT7700 transmission electron microscope (HITACHI, Tokyo, Japan).
Array analysis for mouse cytokines
Expression profiling of cytokines in intestinal epithelium was quantified using Quantibody® Mouse TH17 Cytokine Array 1 (QAM-TH17-1, RayBiotech, Norcross, GA), according to the manufacturer’s instructions. Individual sample was prepared in triplicate for each test. Microarray Analysis Software with GenePix 4400 scanner was used to scan array slides and to acquire fluorescence intensity. The results were analyzed with the RayBio Analysis Tool Excel sheet. Relative concentration of each cytokine was determined using standard curve established in the array and presented as pg/ml.
Hematological parameters
After completing the DSS intervention, all mice were anesthetized. For each mouse, 0.2 ml of whole blood was collected from orbital venous plexus into an EDTA anticoagulation tube. Then, blood routine examination was performed using an automatic hematology analyzer (Mindray BC-2800, Shenzhen, China).
Cell culture and DSS treatment
The human colorectal adenocarcinoma cell line Caco-2 was kindly provided by Cancer Hospital, Chinese Academy of Medical Science, completed STR authentication and mycoplasma detection. Cells were cultured in MEM-EBSS basal medium (Hyclone, Logan, UT, USA) supplemented with 20% heat-inactivated FBS, 1%NEAA, 10 mM HEPES buffer and 1% penicillin/streptomycin in a humidified atmosphere containing 5% CO2 at 37°C. Cells were seeded into 6-well plates at a density of 2 × 105 cells/well. Culture medium was changed every alternative day until fully differentiated into monolayer with 70–80% confluence. Subsequently, Caco-2 cells were treated with DSS at various concentration (5%, 8%, 10%, respectively) for 24 h to establish the injury model.
Cldn-7 shRNA lentiviral transfection
Human Caco-2 cells were transfected with lentivirus as described previously.Citation21 The H1/GFP/Puro lentiviral vectors that target Cldn-7 (Cldn-7 shRNA, sequence: 5ʹ-CTAAGTCCAACTCTTCCAA-3ʹ) or negative control (scrambled shRNA, sequence: 5ʹ-TTCTCCGAACGTGTCACGT-3ʹ) were purchased from Genepharma (Shanghai, China). Transfection of Caco-2 cells was performed according to the manufacturer’s manual. Then, applicable stably transfected cells were screened by puromycin.
Cell viability assay
Caco-2 cells were seeded in 96-well culture plates (Corning) at a density of 5000 cells/well. When reached 70%‐80% confluence, cells were exposed to ±DSS for 24 h. Then, cell viability was measured using Cell Counting Kit-8 (CCK8) kit (Dojindo, Tokyo, Japan). Relative absorbance was measured by a microplate reader at 450 nm. Cell viability (%) was calculated as (OD treated/OD control) x100%.
Detection of cell apoptosis
Flow cytometry was conducted to evaluate apoptosis using PE Annexin V Apoptosis Detection Kit with 7-AAD (BD Biosciences) based on the manufacturer’s instructions. Following DSS treatment, Caco-2 cells were collected and incubated with Annexin V-PE and 7-AAD fluorescence dye for 15 min. Then, cells were analyzed with BD FACScan flow cytometer (BD Bioscience).
Statistical analysis
Data were expressed as mean ± SD. Differences among experimental groups were analyzed using GraphPad Prism 8.0 software (La Jolla, CA, USA). Comparison between two groups was evaluated by the two-tailed unpaired Student’s t-test or Mann-Whitney U test analysis. Differences among three or more groups were analyzed using one-way or two-way ANOVA, followed by a post-hoc test. The significance of Kaplan–Meier survival analysis was calculated by Log-rank test. P < .05 was considered statistically significant.
Declaration of interest statement
The authors declare no competing financial interests.
Author contributions
Lei Ding designed the study, directed the work and approved the final version; Kun Wang performed the experiments, collected and analyzed data and drafted the manuscript; Yuhan Ding and Chang Xu fed the mice; Mengdi Hao and Huimin Li collected clinical tissue samples.
Supplemental Material
Download ()Acknowledgments
We thank Professor Robine for his gift of villin-creERT2 mice and GemPharmatech Co., Ltd for the technical support of model construction.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, Panaccione R, Ghosh S, Wu JCY, Chan FKL, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2018;390(10114):2769–16. doi:10.1016/S0140-6736(17)32448-0.
- Kaplan GG, Ng SC. Understanding and preventing the global increase of inflammatory bowel disease. Gastroenterology. 2017;152(2):313–321.e312. doi:10.1053/j.gastro.2016.10.020.
- Kobayashi T, Siegmund B, Le Berre C, Wei SC, Ferrante M, Shen B, Bernstein CN, Danese S, Peyrin-Biroulet L, Hibi T. Ulcerative colitis. Nat Rev Dis Primers. 2020;6:74.
- Choi CR, Bakir IA, Hart AL, Graham TA. Clonal evolution of colorectal cancer in IBD. Nat Rev Gastroenterol Hepatol. 2017;14:218–229.
- Lasry A, Zinger A, Ben-Neriah Y. Inflammatory networks underlying colorectal cancer. Nat Immunol. 2016;17:230–240.
- Grivennikov SI. Inflammation and colorectal cancer: colitis-associated neoplasia. Semin Immunopathol. 2013;35:229–244.
- Parikh K, Antanaviciute A, Fawkner-Corbett D, Jagielowicz M, Aulicino A, Lagerholm C, Davis S, Kinchen J, Chen HH, Alham NK, et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature. 2019;567:49–55.
- Yu LC. Microbiota dysbiosis and barrier dysfunction in inflammatory bowel disease and colorectal cancers: exploring a common ground hypothesis. J Biomed Sci. 2018;25:79.
- Zihni C, Mills C, Matter K, Balda MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol. 2016;17:564–580.
- Zhu L, Han J, Li L, Wang Y, Li Y, Zhang S. Claudin family participates in the pathogenesis of inflammatory bowel diseases and colitis-associated colorectal cancer. Front Immunol. 2019;10:1441. doi:10.3389/fimmu.2019.01441.
- Gowrikumar S, Ahmad R, Uppada SB, Washington MK, Shi C, Singh AB, Dhawan P. Upregulated claudin-1 expression promotes colitis-associated cancer by promoting β-catenin phosphorylation and activation in Notch/p-AKT-dependent manner. Oncogene. 2019;38(26):5321–5337. doi:10.1038/s41388-019-0795-5.
- Raju P, Shashikanth N, Tsai PY, Pongkorpsakol P, Chanez-Paredes S, Steinhagen PR, Kuo WT, Singh G, Tsukita S, Turner JR. Inactivation of paracellular cation-selective claudin-2 channels attenuates immune-mediated experimental colitis in mice. J Clin Invest. 2020;130(10):5197–5208. doi:10.1172/JCI138697.
- Ding L, Lu Z, Foreman O, Tatum R, Lu Q, Renegar R, Cao J, Chen Y; Ding L, Lu Z, Foreman O, Tatum R, Lu Q, Renegar R, Cao J, Chen YH. Inflammation and disruption of the mucosal architecture in claudin-7–deficient mice. Gastroenterology. 2012;142(2):305–315. doi:10.1053/j.gastro.2011.10.025.
- Tanaka H, Takechi M, Kiyonari H, Shioi G, Tamura A, Tsukita S. Intestinal deletion of Claudin-7 enhances paracellular organic solute flux and initiates colonic inflammation in mice. Gut. 2015;64(10):1529–1538. doi:10.1136/gutjnl-2014-308419.
- Nakayama M, Ishizuka N, Hempstock W, Ikari A, Hayashi H. Na(+)-coupled nutrient cotransport induced luminal negative potential and claudin-15 play an important role in paracellular Na(+) recycling in mouse small intestine. Int J Mol Sci. 2020;21(2):376. doi:10.3390/ijms21020376.
- Luissint AC, Williams HC, Kim W, Flemming S, Azcutia V, Hilgarth RS, Leary MNO, Denning TL, Nusrat A, Parkos CA. Macrophage-dependent neutrophil recruitment is impaired under conditions of increased intestinal permeability in JAM-A-deficient mice. Mucosal Immunol. 2019;12(3):668–678. doi:10.1038/s41385-019-0143-7.
- Mir H, Meena AS, Chaudhry KK, Shukla PK, Gangwar R, Manda B, Padala MK, Shen L, Turner JR, Dietrich P, et al. Occludin deficiency promotes ethanol-induced disruption of colonic epithelial junctions, gut barrier dysfunction and liver damage in mice. Biochim Biophys Acta. 2016;1860(4):765–774. doi:10.1016/j.bbagen.2015.12.013.
- Amasheh S, Fromm M, Günzel D. Claudins of intestine and nephron - a correlation of molecular tight junction structure and barrier function. Acta Physiol (Oxf). 2011;201(1):133–140. doi:10.1111/j.1748-1716.2010.02148.x.
- Singh AB, Uppada SB, Dhawan P. Claudin proteins, outside-in signaling, and carcinogenesis. Pflugers Arch. 2017;469(1):69–75. doi:10.1007/s00424-016-1919-1.
- Garcia-Hernandez V, Quiros M, Nusrat A. Intestinal epithelial claudins: expression and regulation in homeostasis and inflammation. Ann N Y Acad Sci. 2017;1397(1):66–79. doi:10.1111/nyas.13360.
- Wang K, Li T, Xu C, Ding Y, Li W, Ding L. Claudin-7 downregulation induces metastasis and invasion in colorectal cancer via the promotion of epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2017;390(3):797–804. doi:10.1016/j.bbrc.2018.10.049.
- Kuhn S, Koch M, Nübel T, Ladwein M, Antolovic D, Klingbeil P, Hildebrand D, Moldenhauer G, Langbein L, Franke WW, et al. A complex of EpCAM, claudin-7, CD44 variant isoforms, and tetraspanins promotes colorectal cancer progression. Mol Cancer Res. 2020;130(6):553–567. doi:10.1158/1541-7786.MCR-06-0384.
- Darido C, Buchert M, Pannequin J, Bastide P, Zalzali H, Mantamadiotis T, Bourgaux J-F, Garambois V, Jay P, Blache P, et al. Defective claudin-7 regulation by Tcf-4 and Sox-9 disrupts the polarity and increases the tumorigenicity of colorectal cancer cells. Cancer Res. 2012;142(11):4258–4268. doi:10.1158/0008-5472.CAN-07-5805.
- Kim ER. Colorectal cancer in inflammatory bowel disease: the risk, pathogenesis, prevention and diagnosis. World J Gastroenterol. 2014;20(29):9872–9881. doi:10.3748/wjg.v20.i29.9872.
- Zeisel MB, Dhawan P, Baumert TF. Tight junction proteins in gastrointestinal and liver disease. Gut. 2019;68(3):547–561. doi:10.1136/gutjnl-2018-316906.
- Zuo L, Kuo W-T, Turner JR. Tight junctions as targets and effectors of mucosal immune homeostasis. Cell Mol Gastroenterol Hepatol. 2020;10(2):327–340. doi:10.1016/j.jcmgh.2020.04.001.
- Beaugerie L, Svrcek M, Seksik P, Bouvier AM, Simon T, Allez M, Brixi H, Gornet JM, Altwegg R, Beau P, et al. Risk of colorectal high-grade dysplasia and cancer in a prospective observational cohort of patients with inflammatory bowel disease. Gastroenterology. 2013;145(1):166–175.e168. doi:10.1053/j.gastro.2013.03.044.
- Xu C, Wang K, Ding Y-H, Li W-J, Ding L. Claudin-7 gene knockout causes destruction of intestinal structure and animal death in mice. World J Gastroenterol. 2019;25(5):584–599. doi:10.3748/wjg.v25.i5.584.
- Xu C, Wang XN, Li WJ, Wang K, Ding L. Expression and clinical significance of claudin-7 in patients with colorectal cancer. Technol Cancer Res Treat. 2008;68:1–10. doi:10.1177/1533033818817774.
- Quan JC, Peng J, Guan X, Liu Z, Jiang Z, Chen HP, Zhuang M, Wang S, Sun P, Wang HY, et al. Evaluation of clinical significance of claudin 7 and construction of prognostic grading system for stage II colorectal cancer. World J Clin Cases. 2020;8:2190–2200.
- Wen X, Zhao H, Wang L, Wang L, Du G, Guan W, Liu J, Cao X, Jiang X, Tian J, et al. Nobiletin attenuates DSS-induced intestinal barrier damage through the HNF4α-Claudin-7 signaling pathway. J Agric Food Chem. 2020;68:4641–4649.
- Kiesler P, Fuss IJ, Strober W. Experimental models of inflammatory bowel diseases. Cell Mol Gastroenterol Hepatol. 2015;1:154–170.
- Xing T, Benderman LJ, Sabu S, Parker J, Yang J, Lu Q, Ding L, Chen YH. Tight junction protein claudin-7 is essential for intestinal epithelial stem cell self-renewal and differentiation. Cell Mol Gastroenterol Hepatol 2020;9:641–659.
- Bhat AA, Uppada S, Achkar IW, Hashem S, Yadav SK, Shanmugakonar M, Al-Naemi HA, Haris M, Uddin S. Tight junction proteins and signaling pathways in cancer and inflammation: a functional crosstalk. Front Physiol. 2018;9:1942.
- De Robertis M, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L, Signori E, Fazio VM. The AOM/DSS murine model for the study of colon carcinogenesis: from pathways to diagnosis and therapy studies. J Carcinog. 2011;10:9.
- Vermeulen L, Emf DS, Van Der Heijden M, Cameron K, De Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–476.
- Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–850.
- Pouyet L, Roisin-Bouffay C, Clément A, Millet V, Garcia S, Chasson L, Issaly N, Rostan A, Hofman P, Naquet P, et al. Epithelial vanin-1 controls inflammation-driven carcinogenesis in the colitis-associated colon cancer model. Inflamm Bowel Dis. 2010;16:96–104.
- Luo C, Zhang H. The role of proinflammatory pathways in the pathogenesis of colitis-associated colorectal cancer. Mediators Inflamm. 2017;2017:5126048.
- Neal MD, Richardson WM, Sodhi CP, Russo A, Hackam DJ. Intestinal stem cells and their roles during mucosal injury and repair. J Surg Res. 2011;167:1–8.
- Mennillo E, Yang X, Paszek M, Auwerx J, Benner C, Chen S. NCoR1 protects mice from dextran sodium sulfate-induced colitis by guarding colonic crypt cells from luminal insult. Cell Mol Gastroenterol Hepatol. 2020;10:133–147.
- Zhang J, Hou S, Gu J, Tian T, Yuan Q, Jia J, Qin Z, Chen Z. S100A4 promotes colon inflammation and colitis-associated colon tumorigenesis. Oncoimmunology. 2018;7:e1461301.
- Naydenov NG, Feygin A, Wang D, Kuemmerle JF, Harris G, Conti MA, Adelstein RS, Ivanov AI. Nonmuscle Myosin IIA regulates intestinal epithelial barrier in vivo and plays a protective role during experimental colitis. Sci Rep. 2016;6:24161.
- He C, Yu T, Shi Y, Ma C, Yang W, Fang L, Sun M, Wu W, Xiao F, Guo F, et al. 301A promotes intestinal inflammation and colitis-associated cancer development by inhibiting BTG1. Gastroenterology. 2017;152:1434–1448.e1415.
- Chang YY, Yu LC, Yu IS, Jhuang YL, Huang WJ, Yang CY, Jeng YM. Deletion of cadherin-17 enhances intestinal permeability and susceptibility to intestinal tumour formation. J Pathol. 2018;246:289–299.
- Kim Y, Kessler SP, Obery DR, Homer CR, McDonald C. de la Motte CA. Hyaluronan 35kDa treatment protects mice from Citrobacter rodentium infection and induces epithelial tight junction protein ZO-1 in vivo. Matrix Biol. 2017;62:28–39.
- Bian Y, Dong Y, Sun J, Sun M, Hou Q, Lai Y, Zhang B. Protective effect of kaempferol on LPS-induced inflammation and barrier dysfunction in a coculture model of intestinal epithelial cells and intestinal microvascular endothelial cells. J Agric Food Chem. 2020;68:160–167.