
ABSTRACT
Leishmania donovani is the major cause of visceral leishmaniasis (kala-azar), now recognized as the parasitic disease with the highest level of mortality second only to malaria. No human vaccine is currently available. A 36 kDa L. donovani nucleoside hydrolase (LdNH36) surface protein has been previously identified as a potential vaccine candidate antigen. Here we present data on the expression of LdNH36 in Pichia pastoris and its purification at the 20 L scale to establish suitability for future pilot scale manufacturing. To improve efficiency of process development and ensure reproducibility, 4 N-linked glycosylation sites shown to contribute to heterogeneous high-mannose glycosylation were mutated to glutamine residues. The mutant LdNH36 (LdNH36-dg2) was expressed and purified to homogeneity. Size exclusion chromatography and light scattering demonstrated that LdNH36-dg2 existed as a tetramer in solution, similar to the wild-type recombinant L. major nucleoside hydrolase. The amino acid mutations do not affect the tetrameric interface as confirmed by theoretical modeling, and the mutated amino acids are located outside the major immunogenic domain. Immunogenic properties of the LdNH36-dg2 recombinant protein were evaluated in BALB/c mice using formulations that included a synthetic CpG oligodeoxynucleotide, together with a microparticle delivery platform (poly(lactic-co-glycolic acid)). Mice exhibited high levels of IgG1, IgG2a, and IgG2b antibodies that were reactive to both LdNH36-dg2 and LdNH36 wild-type. While the point mutations did affect the hydrolase activity of the enzyme, the IgG antibodies elicited by LdNH36-dg2 were shown to inhibit the hydrolase activity of the wild-type LdNH36. The results indicate that LdNH36-dg2 as expressed in and purified from P. pastoris is suitable for further scale-up, manufacturing, and testing in support of future first-in-humans phase 1 clinical trials.
Introduction
Leishmaniasis is a neglected tropical disease caused by intracellular parasites of the genus Leishmania and transmitted by the bite of phlebotomine sand flies. New estimates from the Global Burden of Disease Study 2013 indicate that the visceral form of leishmaniasis (VL) – also known as kala-azar – causes more than 60,000 deaths annually.Citation1 Therefore, VL is a leading cause of death from parasitic diseases, second only to malaria, and the leading neglected tropical disease in terms of mortality. Most of the world's cases of VL are caused by Leishmania donovani, which is endemic to South Asia, East Africa, and parts of the Middle East.Citation2 There is an urgent need to develop a vaccine for VL, especially in outbreak settings linked to conflict such as the one in southern Sudan during the 1980s and 1990s that resulted in more than 100,000 deaths.Citation2 The economics of introducing a vaccine for VL was recently modeled for Bihar State, India, and it was found that even a modestly protective vaccine was cost-effective.Citation3 Likewise, for cutaneous leishmaniasis (CL), a second major form of the disease, the need and potential economic value of a vaccine has also been demonstrated.Citation4
The 36 kDa nucleoside hydrolase from L. donovani (LdNH36) has been identified as a candidate vaccine antigen and shown to be cross-protective as a DNA vaccine against various Leishmania species causing either VL or CL in mouse and hamster laboratory models.Citation5,6 In contrast to their mammalian hosts, Leishmania protozoan parasites lack the ability for de novo synthesis of purines, which is required in several metabolic processes including nucleic acid synthesis, apoptosis, energy regulation, and signal transduction.Citation7 Leishmania parasites are hence dependent on exogenous purines provided by their host.Citation7,8 In order to metabolize host purine nucleosides, the Leishmania purine salvage pathway utilizes LdNH36 as an essential nucleoside hydrolase.Citation9 The hydrolase has been detected on the cell surface in both promastigote and amastigote forms of L. donovani.Citation10 The surface localization of the hydrolase and the obligatory nature of purine salvage in these parasites, therefore, make the enzyme an attractive antigen for vaccine development. Previous studies have already shown recognition of LdNH36 from sera of patients with VL suggesting LdNH36 has inherent antigenic properties desirable for such a vaccine.Citation11
LdNH36 as a DNA vaccine has been shown to be efficacious in a mouse model of VL;Citation12 however, DNA vaccines have so far not advanced to licensure in humans.Citation13,14 Accordingly, we are exploring the feasibility of developing LdNH36 as a recombinant protein-based vaccine. For vaccine product development to be technically and economically feasible, the establishment of a simple, scalable process that produces sufficient quantities of high-purity recombinant protein antigen for both clinical testing and later industrial scale manufacture is of paramount importance.
An Escherichia coli expression system has been previously used to produce a recombinant LdNH36 protein with a maltose binding tag.Citation15 While E. coli can be tapped as a robust expression system to support manufacturing processes via high expression yields, it also typically generates high levels of contaminants such as host cell proteins, endotoxin, and DNA, which can exacerbate the downstream purification process.Citation16 The Pichia pastoris yeast expression system, on the other hand, offers expression of a secreted protein product, which has no inherent endotoxin present and low levels of DNA, host cell proteins, and other cellular contaminants. This can obviate the need for a tag and ultimately allow for a more efficient, and thus less costly, scalable and reproducible purification process.Citation17-19 The eukaryote P. pastoris also provides a more consistent expression platform in terms of folding and post-translational modifications with the native parasite-expressed LdNH36; however, the potential for hyperglycosylation leading to heterogeneous high-mannose glycoforms of the recombinant protein, which can adversely affect the ability to produce a consistent, well-characterized, purified protein, must be considered.Citation20 This concern can be mitigated using genetic engineering to modify putative N-glycosylation sites via asparagine (N)-to-glutamine (Q) mutations, an approach used previously in protein vaccine development.Citation21-24
The immunogenic properties of the yeast-expressed, engineered LdNH36 (LdNH36-dg2) were evaluated in BALB/c mice. As recombinant proteins alone often elicit weak immune responses, especially T helper 1 (TH1) immune responses such as those found necessary for protective immunity against intracellular parasites, including Leishmania,Citation25 we are evaluating LdNH36-dg2 as an immunogen using innovative delivery platforms, novel adjuvants, or both. With respect to the former, LdNH36-dg2 was formulated in a poly(lactic-co-glycolic acid) (PLGA) microparticle delivery platform, which has been shown to enhance the overall immune response by facilitating the uptake of antigens by antigen-presenting cells.Citation26-28 In order to augment TH1-type immunity, the vaccine particles were supplemented with CpG oligodeoxynucleotide (CpG) adjuvant in order to trigger the cellular pathway mediated by the Toll-like receptor 9 (TLR9). CpG has shown promise of regulatory acceptance in Phase I – III clinical trials and has been found to improve protection against L. major challenge.Citation29-31
In this study, we engineered N-linked glycosylation sites on a P. pastoris-expressed LdNH36 recombinant protein antigen (LdNH36-dg2) and developed a scalable expression and purification process. We also conducted an animal study to evaluate the immune responses of LdNH36-dg2 when formulated in PLGA microparticles, with or without the presence of CpG adjuvant, by measuring antigen-specific TH1-related IgG2a and TH2-related IgG1 antibody titers.Citation32
Results
Engineering of LdNH36 expressed in P. pastoris X-33 reduced hyperglycosylation
Small scale cultures (10 mL), induced with 0.5% methanol for 72 h, showed that P. pastoris-expressed wild-type LdNH36 (LdNH36-Y-WT) was detectable by anti-LdNH36 mouse sera generated against wild-type his-tagged, E. coli-expressed LdNH36 (LdNH36-E-WT). Lanes 1–3 in show different LdNH36 recombinant proteins expressed in P. pastoris, while lane 4 shows the E. coli expressed protein. The molecular weight (MW) of LdNH36-Y-WT (lane 1) was significantly higher than the E. coli expressed protein (lane 4). LdNH36-Y-WT also showed a pronounced high molecular weight (HMW) smear not present in LdNH36-E-WT, suggesting the yeast expression system caused the presence of heterogeneous high-mannose glycoforms. After mutating 4 of the N-linked glycosylation sites from asparagines to serines (LdNH36-dg), the molecular weight of the main band was decreased and the HMW smear was reduced (lane 2), confirming that hyperglycosylation was indeed an issue in our system. Additionally, mutating asparagines to glutamines (LdNH36-dg2) resulted in a main band (Lane 3) without a HMW smear that migrated with an apparent molecular weight similar to the non-glycosylated E. coli expressed protein (Lane 4). Unlike LdNH36-E-WT, which migrated as one band (see Fig. S1), a minor second band was observed migrating just above the main band for LdNH36-dg2. Although the 2 remaining asparagines (N160 and N181) in LdNH36-dg2 with the N-glycosylation consensus sequence (N-X-S/T) had a prediction value below the threshold by NetNGlyc 1.0 Server, glycosylation of the sites is possible, which could lead to the slight increase in MW. O-linked glycosylation, which can also occur in P. pastoris, could be another source of the increased MW. The glutamine mutations also resulted in increased expression levels of LdNH36 compared to the serine mutations. Thus, the LdNH36-dg2 construct with the mutations detailed in was selected for further scale-up and immunogenicity testing.
Figure 1. Expression of LdNH36 constructs in P. pastoris X-33 compared with E. coli-expressed wild-type LdNH36, evaluated by Western blot with anti-LdNH36 mouse sera (reduced 4–20% Tris-glycine gel/chemiluminescence detection). Lane 1: 10 µL of wild-type LdNH36 culture supernatant expressed in P. pastoris; Lane 2: 10 µL of LdNH36-dg culture supernatant expressed in P. pastoris; Lane 3: 10 µL of LdNH36-dg2 culture supernatant expressed in P. pastoris; Lane 4: 50 ng (determined by 280 nm absorbance) of purified, His-tagged wild-type LdNH36 expressed in E. coli as control.
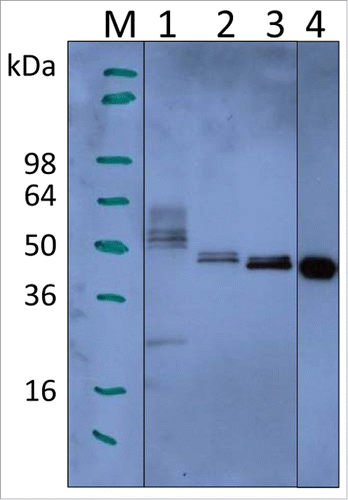
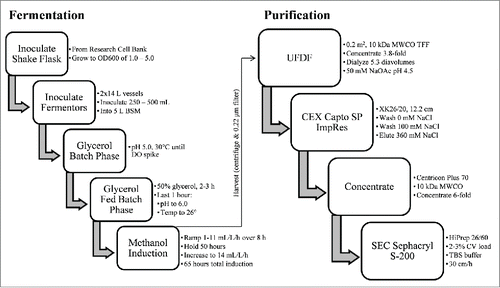
Figure 3. Schematic of LdNH36-dg2 manufacturing compatible fermentation and purification process.
Scalable fermentation process yields 1.2 g of LdNH36-dg2
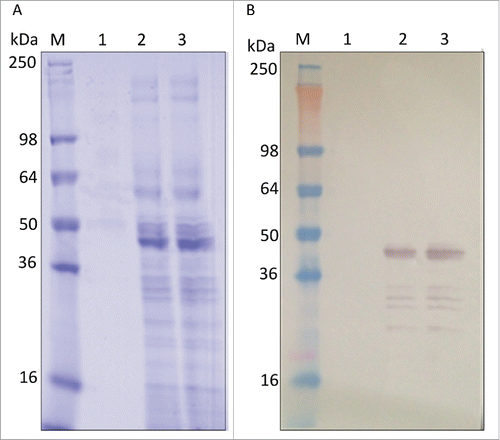
A scalable fermentation process was developed using standard bioprocess conditions with the intent to facilitate future transfer to the industrial scale. The process outlined in yielded 1.2 g of unpurified LdNH36-dg2 from two 10 L runs. The dissolved oxygen (DO) trace and oxygen feed rate were monitored during the fermentation, and both were consistent with healthy, metabolically active cells during pre- and post-induction phases. Different induction times were evaluated, and the optimized duration of 72 h was selected since additional time did not result in increased LdNH36-dg2 (). This relatively short duration is also desirable for future manufacturing.
Figure 2. Amino acid sequence of LdNH36-dg2. The 4 N-glycosylation site mutations (N!Q) are underlined. None of the mutation sites are present in F3 region (bold), which showed highest protection in studies by Nico et al., 2010.

Figure 4. Fermentation results with reduced 4–20% Tris-glycine gels. (A) SDS-PAGE with Coomassie Blue staining and (B) Western blot with anti-LdNH36/colorimetric detection. Lane M: molecular weight marker; Lane 1: pre-induction; Lane 2: 72 h of induction; Lane 3: 96 h of induction.
Scalable purification process yields ∼0.5 g of homogenous LdNH36-dg2
A simple, 2-step column purification process was developed to be performed at room temperature using standard bioprocess conditions that will allow ease and decreased cost upon scale-up and manufacturing. The finalized process outlined in was performed on fermentation supernatant from two 10 L fermentations combined into one “20 L” purification batch. The process first utilized a TFF system with a 10 kDa molecular weight cut-off (MWCO) cassette to buffer exchange the P. pastoris fermentation supernatant and decrease the pH. The 10 kDa MWCO, which is significantly lower than the molecular weight of LdNH36, showed no detectable loss of LdNH36-dg2. Subsequent loading onto a Capto SP ImpRes column at pH 4.5 captured LdNH36-dg2, while allowing the majority of contaminants to flow-through. A 100 mM NaCl wash removed loosely bound contaminants, and LdNH36-dg2 eluted in 360 mM NaCl with a recovery of 69% and a significant increase in purity (, lanes 2 vs. 3). Prior to loading onto the Sephacryl S-200 HR size-exclusion chromatography column (SEC200), the SP pool was concentrated with spin cartridges to reduce the number of SEC200 cycles. A 10 kDa MWCO was sufficient to recover 75% of LdNH36-dg2. Superdex 200 chromatography removed the majority of the remaining higher and lower molecular weight species (, lanes 4 vs. 5/6) with a 72% recovery. The individual step yields and purity are outlined in . The overall process produced 454 mg (37% overall recovery) of LdNH36-dg2 from the two 10 L fermentations and provides a scalable approach to produce LdNH36-dg2 for the intended clinical trials.
Figure 5. In-process sample analysis with 4–12% Bis-Tris gels. (A) SDS-PAGE with Coomassie blue staining, (B) Western blot with anti-LdNH36/colorimetric detection), and (C) HCP Western blot with anti-P. pastoris/colorimetric detection. Lanes 1–6 are non-reduced and lanes 7–8 are reduced. Lanes M: molecular weight standard; Lane 1: fermentation supernatant; Lane 2: post-TFF; Lane 3: Capto SP pool; Lane 4: concentrated Capto SP pool; Lane 5 and 8: SEC200 pool low load; Lane 6 and 7: SEC200 pool high load.
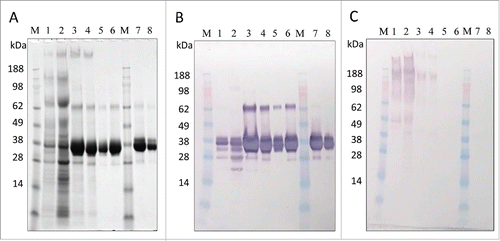
Table 1. Purification table from 20 L run SDS-PAGE densitometry.
Characterization of purified LdNH36-dg2 protein shows high purity and consistency with crystallized L. major nucleoside hydrolase
Analysis by SDS-PAGE, HPLC-SEC, and Western Blot showed increased purity throughout the purification process, resulting in a highly enriched final product. SDS-PAGE densitometry analysis ( and ) demonstrated an increase in purity from 24% to 97% throughout the process. The main band by SDS-PAGE (, lanes 5–8) migrated near 36 kDa and was immunoreactive to wild-type anti-LdNH36 sera, which was generated in mice using E.coli recombinant wild-type his-tagged LdNH36. The 2 other visible bands in the purified LdNH36-dg2 (, lanes 5–8), which migrated near 26 kDa and 64 kDa, were also immunoreactive and comprised < 0.7% and 1.5% of the total lane density, respectively. Host cell protein (HCP) reactivity against P. pastoris was visible in the initial fermentation supernatant (FS) and was greatly reduced after capture by Capto SP (, lanes 1 vs. 3). After SEC200, no HCP reactivity was observed.
LC-MSE peptide mapping was used as an orthogonal method to identify proteins present in the main 36 kDa and minor 64 kDa bands. The main 36 kDa band was composed of 100% LdNH36-dg2, which was consistent with both (anti-LdNH36 and anti-HCP) Western blot results, while the minor 64 kDa band was composed of 88% P. pastoris proteins and 12% LdNH36-dg2, which was inconsistent with the HCP Western blot results. The Western blot method to detect HCPs can be less sensitive than LC-MSE as it relies on polyclonal antibody sera generated against several proteins simultaneously. Thus, combining the LC-MSE and SDS-PAGE data, the final purified product consisted of > 97% LdNH36-dg2.
LC-MSE peptide mapping was also used to evaluate the presence of glycosylation. To obtain full coverage of the amino acid sequence, both a trypsin and Endo Glu-C digestion was performed. No O-linked glycosylation was observed. Of the 2 remaining N-linked glycosylation consensus sequences (N-X-S/T), no glycosylation was observed for site N160, but glycosylation was observed for some of the peptides at site N181 (< 20%), as shown in Fig. S3. A PNGase F digestion was performed confirming that the top band observed in the reduced LdNH36-dg2 () was a result of glycosylation (Fig. S2).
HPLC-SEC analysis in showed improvement in purity from < 7% to 90%. LdNH36-dg2 is present as one main peak at 132 kDa, which is consistent with the formation of a native tetramer as previously characterized by Shi et al. for the L. major nucleoside hydrolase having 95% homology to LdNH36.Citation33
Figure 6. HPLC-SEC and DLS characterization of LdNH36-dg2. (A) HPLC-SEC chromatographs of in-process samples are shown with the molecular weight standard in the top chromatograph and corresponding MW of peaks labeled in kDa. LdNH36-dg2 is present at retention time of 30 minutes corresponding to a MW of 132 kDa, demonstrating that LdNH36-dg2 is a tetramer in solution. The predicted structure of the molecule as a tetramer is shown in (B) with the mutated glutamines highlighted in red. (C) DLS results of the purified LdNH36-dg2 (SEC200 Pool) are presented as % intensity and demonstrate a similar MW (135 kDa). The polydispersity is 13.2%, indicating a monodisperse purified LdNH36-dg2.
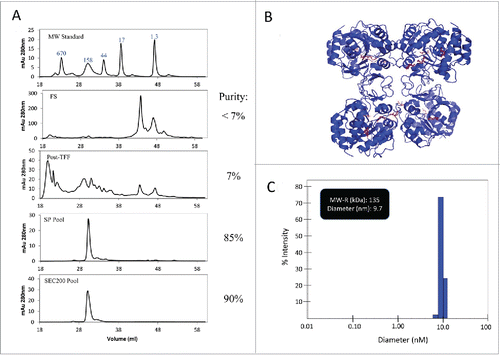
A predicted structural model of the LdNH36-dg2 tetramer is shown in . The tetramer assembly is likely conserved across both species, and most of the residues at the interface (identified in PDBsumCitation34) are identical. The only difference is a conservative histidine to asparagine substitution at position 233, which forms a non-bonded contact via the conserved beta carbon (CB). Additionally, none of the 4 glutamine to asparagine mutations are present at the interface.
Dynamic light scattering, as shown in , also confirmed that the recombinant protein formed a native tetramer with a molecular weight near 135 kDa (hydrodynamic radius of 4.85 nm). Only one main species was present, suggesting the removal of the higher and lower molecular weight species. A polydispersity of 13.2% demonstrated a monodisperse final product.Citation35
Characterization of PLGA microparticles containing LdNH36-dg2 and CpG
The formulation of LdNH36-dg2 with CpG-containing microparticles resulted in microparticle mass yields of approximately 50%, with loading levels of 21.6 – 22.3 µg LdNH36-dg2 protein (108 – 112% efficiency) and 36.9 µg CpG (92.2% efficiency) per mg of particles. SEM images of LdNH36-dg2-loaded, CpG-loaded, and empty microparticles were generated prior to lyophilization and are shown in .
Figure 7. Scanning electron microscope (SEM) images of LdNH36-dg2- or CpG-loaded microparticles or empty microparticles. LdNH36-dg2 protein was encapsulated in poly(lactic-co-glycolic acid) (PLGA) microparticles using a water-oil-water double emulsion method, CpG oligonucleotide adjuvant was encapsulated using an oil-water emulsion method preceded by ion-pairing, and empty PLGA microparticles were prepared by an oil-water emulsion method.

Serum antibody response
All mice vaccinated with LdNH36-dg2-containing formulations generated substantial titers of serum antibodies of the subclasses IgG1, IgG2a, and IgG2b when using LdNH36-dg2 as the coating antigen, as shown in . For the IgG1 subclass, all groups vaccinated with microparticle-formulated LdNH36-dg2 (MP/LdNH36-dg2), with or without CpG adjuvant, had at least 10-fold higher geometric mean titers than the non-microparticle (soluble) LdNH36-dg2/CpG group (p < 0.001); however, there were no significant differences in IgG1 mean titers among the MP/LdNH36-dg2 groups, regardless of the presence or dosage of CpG. For the IgG2a and IgG2b subclasses, the differences between vaccinated groups were less pronounced; however, both IgG2 subtypes were consistently higher when the antigen was delivered with a microparticle and in the presence of CpG. There was a consistent trend of higher titers with the use of microparticles and with increasing doses of CpG. For IgG2a, the MP/LdNH36-dg2/CpG (80 μg) vaccine elicited at least 25-fold higher mean titers than the soluble LdNH36-dg2/CpG (20 μg) and the MP/LdNH36-dg2 vaccine (p < 0.05). For IgG2b, the MP/LdNH36-dg2/CpG (20, 40, and 80 μg) vaccines had 14- to 45-fold higher mean titers than the soluble LdNH36-dg2/CpG (20 μg) vaccine (p < 0.01), and the MP/LdNH36-dg2/CpG (80 μg) vaccine had 10-fold higher mean titers than the MP/LdNH36-dg2 vaccine (p < 0.05).
Figure 8. Serum antibody titers to (A) LdNH36-dg2 and (B) LdNH36-E-WT (non-polyhistidine tagged) for subtypes IgG1, IgG2a, and IgG2b in response to vaccination with microparticle-formulated LdNH36-dg2 protein and CpG or control formulations. BALB/c mice were vaccinated subcutaneously at weeks 0 and 3 with 40 μg of LdNH36-dg2 protein plus varying amounts of CpG adjuvant (10, 20, 40, or 80 μg) encapsulated in separate PLGA microparticles (MP). Vaccine controls included microparticle-LdNH36-dg2 and soluble LdNH36-dg2 + CpG. Mice were vaccinated in 2 cohorts, separated by 2 d (cohort 1, filled triangles; cohort 2, open squares). Week 5 serum was analyzed by ELISA for LdNH36-dg2- and LdNH36-E-WT-specific IgG1, IgG2a, and IgG2b responses. Negative control groups (microparticle-CpG, empty microparticle, and PBS) had antibody titers less than 103 and are not plotted. *p < 0.05, **p < 0.01, ***p < 0.001.
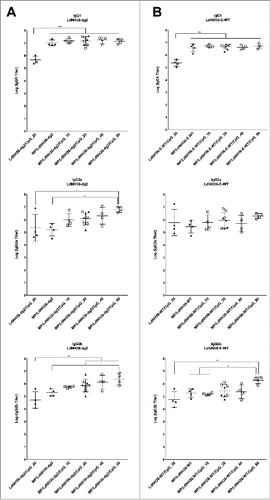
In addition, all mice vaccinated with LdNH36-dg2-containing formulations generated substantial titers of serum antibodies of the subclasses IgG1, IgG2a, and IgG2b when using LdNH36-E-WT as the coating antigen, as shown in . All groups vaccinated with microparticle-formulated LdNH36-dg2 (MP/LdNH36-dg2), had significantly higher IgG1 geometric mean titers than the non-microparticle (soluble) LdNH36-dg2/CpG group (p < 0.001). For IgG2a subclass, there were no significant differences, regardless of the presence of microparticles or dosage of CpG. For IgG2b, the MP/LdNH36-dg2/CpG (80 μg) vaccines had significant higher mean titers than the soluble LdNH36-dg2/CpG (20 μg) vaccine (p < 0.01), MP/LdNH36-dg2 vaccine (p < 0.05) and MP/LdNH36-dg2/CpG (10 μg) vaccine (p < 0.05).
The titers of serum antibodies detecting LdNH36-dg2 were significantly higher than those detecting LdNH36-E-WT for the IgG1 (p < 0.0001) and IgG2b (p < 0.05) subtypes; however, there was no significant difference for the IgG2a subtype. For IgG1, the differences between the antibody titers were within a 95% confidence interval of 2.11- to 3.80-fold; for IgG2b, the differences were within a 95% confidence interval of 1.07- to 3.95-fold.
The negative control groups (microparticle-CpG, empty microparticle, and PBS) all had negligible antibody responses against LdNH36-dg2 and LdNH36-E-WT (individual titers < 103) and thus were excluded from the data plots and statistical analysis.
Antibody inhibition assay
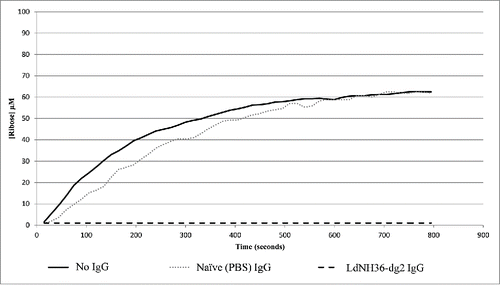
While LdNH36-E-WT was able to hydrolyze inosine, LdNH36-dg2 was not. The N39 point mutation is most likely the cause of the activity loss because it was shown to be involved in substrate recognition.Citation33 Since activity of the antigen is not a pre-requisite for protective efficacy, we further evaluated if the antibodies generated by LdNH36-dg2 could inhibit the activity of the wild type, which can, in some cases, confer protection. While IgG purified from mice injected with PBS showed no inhibition of hydrolase activity, IgG purified from mice injected with LdNH36-dg2 showed complete inhibition of hydrolase activity.().
Figure 9. Antibody Inhibition of LdNH36-E-WT (non-polyhistidine tagged) Nucleoside Hydrolase Activity. LdNH36-E-WT alone shows hydrolysis of inosine to ribose that proceeds at a similar rate when purified IgG from sera of mice injected with PBS (naïve mice) is added to the reaction. Addition of purified IgG from sera of mice injected with LdNH36-dg2 completely inhibits the hydrolase activity.
Discussion
LdNH36, the L. donovani nucleoside hydrolase, is being developed as a candidate recombinant antigen for a leishmaniasis vaccine. In this work, we report the expression of the LdNH36 antigen as a secreted protein in P. pastoris. The P. pastoris expression system provides superior benefits to the E. coli expression systems, including secretion of the target protein as a soluble and stable recombinant protein into the fermentation media.Citation17,18 The fermentation media, which has minimal cell-derived contaminants including endotoxin and DNA, can be taken directly into purification, allowing for streamlined downstream and quality control development without the need for an encoded purification tag. This advantage can reduce the vaccine production cost, which is an important consideration for a leishmaniasis vaccine that targets a major disease of poverty worldwide. Economic modeling has shown that a cost of ≤ US $2 per dose for a preventive vaccine would be economically advantageous in comparison to currently available therapeutic regimens.Citation4 Elimination of expensive process components, such as purification tags and complex purification schemes, can serve as a useful guide toward that goal.
Wild-type LdNH36 was expressed as a secreted protein in P. pastoris, although Western blot analysis suggested the presence of heterogeneous high-mannose type glycosylation, commonly seen in yeast systems.Citation20 To enable batch-to-batch glycoform consistency and to circumvent potential antigenic dampening, point mutations were introduced in 4 putative N-linked glycosylation sites in order to produce a recombinant protein with minimal glycosylation. This approach has been taken previously in order to reduce the heterogeneous glycosylation in recombinant protein vaccine candidates, including severe acute respiratory syndrome (SARS) spike protein receptor binding domain, malaria PfAMA-1, hepatitis C E1, and simian immunodeficiency virus SIVmac239.Citation21-24,36 Initial mutation of the 4 asparagine residues to serines resulted in a significant reduction in the observed heterogeneous hyperglycosylation, while subsequent mutations of the 4 asparagines to glutamines had the added advantage of improving the yields. The improvement in yield may be related to glutamine being closer in physicochemical properties to asparagine than serine. The LdNH36-dg2 protein migrated as 1 major band with a second faint band immediately above it on a reduced SDS-PAGE gel. LC-MSE peptide mapping and PNGase F digestion confirmed the shift in MW of the second band was a result of a small portion (< 20%) of the peptides being glycosylated on one of the 2 remaining N-linked glycosylation consensus sequences (N180). During development, this 2-band pattern was reproducibly observed, which allowed for conformation of lot-to-lot consistency. Since the native protein, as discussed below is a tetramer, separating the monomeric bands to evaluate differences in immunogenicity or efficacy would be difficult. Both mutants were immunoreactive against anti-sera generated from wild-type LdNH36 expressed in E.coli, demonstrating that the mutations did not affect antibody recognition (antigenicity). Moreover, the locations of the point mutations are outside of the F3 region of LdNH36, which is considered to be among the most antigenic regions of the molecule (see ).Citation37 In addition, it had been previously reported that intact carbohydrate epitopes were not required for recognition by anti-LdNH36 serum generated against LdNH36 purified directly from L. donovani (LdNH36 is referred to as GP36 in that report).Citation38
Using the optimized LdNH36-dg2 construct, a fermentation process was developed and executed at the 2 × 10 L scale. The equipment and reagents are commonly found in manufacturing and will allow for a simple transfer to a cGMP manufacturing campaign. High expression levels were observed with LdNH36-dg2 as the main species, allowing for a simple and efficient purification process. We developed a 2-step chromatography purification method, without a polyhistidine (or other) tag, that also utilizes standard bioprocess equipment and reagents. The supernatant was separated by centrifugation and concentrated/dialyzed with a tangential flow filtration system. This facilitated capture by cation exchange chromatography performed at 1.5 pH units below the predicted pI of LdNH36-dg2 (6.06 ExPASY ProtParam toolCitation39) allowing an increase in purity from 24% to 89%. Subsequent size exclusion chromatography removed remaining low and high molecular weight species to yield ∼0.5 g of highly purified LdNH36-dg2 protein. By maintaining individual step recoveries of 69% or greater and by limiting the number of process steps, the achieved overall recovery (37%) enables sufficient LdNH36-dg2 to be produced from a 20 L cGMP manufacturing campaign to support both phase 1 clinical trials and subsequent 5-y stability testing.
Combining the results of SDS-PAGE, Western blots (anti-LdNH36 and anti-HCP), HPLC-SEC, and LC-MSE peptide mapping, the final purified LdNH36-dg2 consisted of > 97% LdNH36-dg2 with > 90% present as a consistent species. The purified LdNH36-dg2 had the expected native tetramer confirmation, as confirmed by HPLC-SEC and DLS, consistent with the L. major nucleoside hydrolase crystal structure.Citation33
To confirm the immunogenic properties of LdNH36-dg2, an immunogenicity study was conducted in mice. LdNH36-dg2 purified protein was formulated both with and without CpG adjuvant in a microparticle-based delivery system. The PLGA microparticle formulation was utilized to enhance the overall immune response to the protein,Citation26,27 and the CpG adjuvant was utilized to bias the immune response toward a TH1 type.Citation29 We observed that all groups given LdNH36-dg2-containing vaccines generated substantial serum IgG antibody reactivity to both LdNH36-dg2 and LdNH36-E-WT, indicating a strong immunogenic potential of the recombinant LdNH36-dg2 protein (). The use of a PLGA microparticle delivery system resulted in consistently higher IgG1 titers in response to both LdNH36-dg2 and LdNH36-E-WT coated plates that were independent of the presence or dosage of CpG adjuvant. This effect of the microparticle delivery system is consistent with the general trend reported in the literature, which shows enhancement of antibody responses to protein antigens.Citation26,27 The use of CpG adjuvant in the microparticle formulation shows a clear trend of increasing IgG2a and IgG2b titers with increasing CpG dosage in response to LdNH36-dg2 coated plates, but such a trend was not statistically significant in response to LdNH36-E-WT coated plates. This result of serum antibody titers to LdNH36-dg2 is consistent with other reported studies in which adding CpG to protein antigen elicited higher IgG2a and IgG2b titers in BALB/c mice.Citation29,40,41 Although the IgG1 and IgG2b titers in response to LdNH36-E-WT were lower than to LdNH36-dg2, all microparticle-formulated groups showed IgG1 titers greater than 106 and IgG2b titers greater than 105. The IgG2a titers in response to LdNH36-E-WT and LdNH36-dg2 were not significantly different, and they were both greater than 105 regardless of the formulations. As titers of IgG2a and IgG2b are associated with a TH1-type immune response in BALB/c mice, which has been shown to correlate with protection against Leishmania infection, LdNH36-dg2 shows potential promise as a protective vaccine.Citation32
While the point mutations affected the hydrolase activity of LdNH36-dg2, the mutant protein was still able to elicit IgG antibodies capable of neutralizing the activity of the wild type protein (LdNH36-E-WT). This data suggests the potential for protective properties of an LdNH36-dg2 vaccine that will need to be confirmed in a challenge model.
In summary, LdNH36 was expressed in P. pastoris with specific amino acid point mutations introduced in the genetic construct to reduce the heterogeneity of the resulting glycoforms, which facilitated process development and quality control. A fermentation and purification process was developed and demonstrated up to a 20 L scale, which resulted in ∼0.5 g of LdNH36-dg2 with a purity of > 97%, suitable for production at the manufacturing scale to support future first-in-humans phase 1 clinical trials. Immunogenicity of LdNH36-dg2 was demonstrated using a PLGA microparticle-based system with and without CpG adjuvant. The use of PLGA microparticles resulted in consistently higher IgG1 responses regardless of the presence of CpG adjuvant although the presence of CpG adjuvant did result in enhanced IgG2a and IgG2b levels (TH1-associated), which has been shown in murine systems to correlate with protection. This data suggests that the recombinant LdNH36-dg2 has the potential to be a successful candidate vaccine. Further studies will be conducted in animals to validate this work in an efficacy model and transfer the process to a cGMP manufacturer for eventual phase 1 clinical trials.
Methods
Expression of mutated LdNH36 in P. pastoris X-33
A wild-type (LdNH36-Y-WT) and 2 mutant constructs (LdNH36-dg and LdNH36-dg2) of LdNH36 (Genbank: XP_003860171.1) were generated for expression in P. pastoris X-33. Site-directed mutagenesis was performed in an attempt to reduce the hyperglycosylation of LdNH36-Y-WT expressed in yeast. The DNA coding for 4 asparagine residues (N39, N77, N89 and N185) at predicted N-glycosylation sites (NetNGlyc 1.0 ServerCitation42) were mutated. The first mutant construct, LdNH36-dg, exchanged the 4 asparagine residues for serine residues, and the second mutant construct, LdNH36-dg2, exchanged the 4 asparagine residues for glutamine residues.
The genetically engineered DNA sequence for all 3 constructs was codon optimized based on yeast codon preference, synthesized by GeneScript (Piscataway, NJ) and cloned into pUC57. PCR amplified coding DNA was gel purified and ligated into the yeast expression vector pPICZαA (Life Technologies; Grand Island, NY) using EcoRI and XbaI restriction sites. Ligated vectors were transformed into E.coli DH5α competent cells (Life Technologies; Grand Island, NY), and clones were selected on low salt LB plates with Zeocin. Plasmid DNA was extracted with Qiagen Miniprep plasmid extraction kit (Valencia, CA) and verified by DNA sequencing (LoneStar Labs, Houston, TX). The sequence confirmed recombinant plasmid (LdNH36/pPICZαA) was transformed into P. pastoris X-33 by electroporation after being linearized by SacI digestion. Transformants were selected on Zeocin containing plates. The expression of recombinant LdNH36 was induced with 0.5% methanol in 10 mL shake tube cultures, and the clone with the highest expression yield was selected to generate a research seed stock.
Fermentation of LdNH36-dg2
Fermentation was conducted in 2 Eppendorf (Hauppauge, NY) Celligen 310 bioreactors with 14 L vessels and the Eppendorf BioCommand software. To initiate the process, 1 L of seed culture was prepared for inoculation of both vessels. The glycerol research seed stock of LdNH36-dg2 (1 mL) was added to 1 L of buffered minimal glycerol (BMG; as per Stratton but referred to as MGY mediumCitation43) in a shake flask and incubated at 29 ± 1°C with agitation at 250 ± 10 rpm overnight. At an OD600 of 1.52, 450 mL of the seed culture was aseptically transferred into each vessel for a final OD600 value of 0.14. Each vessel contained 5 L of sterile basal salt media (BSM) as per StrattonCitation43 and 4.5 mL of PTM4 trace salts as per StrattonCitation43 modified with 4.5 mL/L of 0.02% d-biotin. Prior to inoculation, the pH was adjusted to 5.00 ± 0.2. During the batch phase, the pH was maintained at this level with 14% ammonium hydroxide. Fermentation set points also included temperature at 30±1°C, dissolved oxygen at 30%, air flow at 0.5–1.0 standard liters per minute (SPLM), and agitation at 500 rpm. Antifoam 204 (Sigma-Aldrich; St. Louis, MO) was added as needed to minimize foaming.
A dissolved oxygen (DO) spike was observed approximately 22 h after inoculation indicating depletion of glycerol. Following the spike, a fed-batch was initiated with 50% glycerol feed at 15 mL/L/h for approximately 2–3 h. During the last hour, the pH was shifted from 5.0 to 6.0, and the temperature was shifted from 30°C to 26°C. Pre-induction samples were taken at this point.
After the fed-batch, methanol induction was initiated with an initial 100% methanol bolus for 5 min followed by a 0 to 100% methanol ramp from 1 mL/L/h to 11 mL/L/h over an 8 h period. The 100% methanol included 12 mL/L PTM4 trace salts and 12 mL/L 0.02% biotin. The methanol flow rate was maintained at 11 mL/L/h for 50 h until the oxygen demand went up in both cultures. At that point, the flow rate was increased to 14 mL/L/h, which maintained adequate oxygen control. After 65 h of induction, the culture was harvested, and the supernatant was separated by centrifugation in 1 L bottles at 12,200 × g for 45 min using an Avanti J-26 XPI (Beckman Coulter; Brea, CA) set at 4°C. Following 0.22 µm Corning PES bottle top vacuum filtration, approximately 11.5 L of fermentation supernatant (FS) was obtained.
Purification of LdNH36-dg2
All purification steps were performed at room temperature except the post-cation exchange chromatography (CEX) concentration step, which was performed at 4°C due to the centrifugation. The supernatant from both 10 L fermentations was combined (11.5 L of Fermentation Supernatant (FS) total) and concentrated 3.8-fold followed by dialysis against 50 mM sodium acetate (NaOAc) pH 4.5. The duration was approximately 4 h with 3 Millipore (Billerica, MA) Pellicon 2 mini Biomax-10 A ultrafiltration cassettes (10 kDa molecular weight cutoff (MWCO)). The dialysis required 5.3 diavolumes, and the overall transmembrane pressure (TMP) was 8.1 psi with a flux of 21.4 L/m2/h. The concentrated/dialyzed FS (3.26 L) was then loaded in 3 cycles on an XK26, 12.2 cm Capto SP ImpRes column (GE Healthcare) at 180 cm/h using an AKTA Avant 150 (GE Healthcare). Two wash steps were performed using the dialysis buffer and 50 mM NaOAc pH 4.5, 100 mM sodium chloride (NaCl) followed by an elution with 50 mM NaOAc pH 4.5, 360 mM NaCl. The Capto SP elution pools were combined and concentrated 6-fold using 4 Millipore Centricon Plus 70 spin cartridges (10 kDa MWCO). Finally, the concentrated Capto SP pool was divided into 11 batches (9 equal to 2.9% of the column volume (CV) and the remaining 2 at 2.3% and 2.0% of CV) and loaded in 11 cycles onto a Sephacryl S-200 HR HiPrep 26/60 column (SEC200) (GE Healthcare) at 30 cm/h using the AKTA Avant 150. LdNH36-dg2 was eluted using 30 mM Tris pH 7.5, 150 mM NaCl (TBS). The main peaks from all cycles were pooled to create the final purified LdNH36-dg2 pool.
SDS-PAGE and Western blotting
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed with Life Technologies pre-cast Tris-glycine and Bis-Tris gels according to manufacturer's instructions. Coomassie Blue (GE Healthcare Phastgel Blue R-350), β-mercaptoethanol (Sigma-Aldrich; St. Louis, MO), and SeeBlue Plus 2 (Life Technologies) were used as the staining agent, reducing agent, and molecular weight standard, respectively. Gels were imaged and analyzed with a GE ImageScannerIII and Labscan/ImageQuant TL software. For SDS-PAGE quantitation, a reduced 12% Bis-Tris gel (Life Technologies) was performed on samples diluted within the range of a bovine serum albumin (BSA) calibration curve (0.20 µg to 0.92 µg per well).
Western blot analysis was performed by probing with anti-LdNH36 mouse polyclonal sera produced in-house from purified, E.coli expressed, wild-type his-tagged recombinant LdNH36 primary antibody. Blots using chemiluminescence detection (Amersham ECL Prime Western Blotting Detection Reagent; GE Healthcare) were transferred to a polyvinylidene fluoride (PVDF) membrane and probed with a 1:3,000 dilution of primary antibody and a 1:5,000 dilution of secondary antibody (anti-mouse horseradish peroxidase labeled IgG; Life Technologies). Blots using colorimetric detection (5-bromo-4-chloro-3′-indolyphophate/nitro-blue tetrazolium (BCIP-NBT) (KPL; Gaithersburg, MD) were transferred to a nitrocellulose membrane and probed with a 1:4,000 dilution of primary antibody and a 1:1,000 dilution of secondary antibody (anti-mouse alkaline phosphatase labeled IgG; KPL).
Host cell protein (HCP) detection
Presence of host cell proteins was evaluated by transferring the proteins from a 4–12% Bis-Tris SDS-PAGE gel (performed as described above) onto a nitrocellulose membrane and probing with a 1:1,000 dilution of a goat anti-P. pastoris primary antibody (Cygnus Technologies; Southport, NC) followed by a 1:5,000 dilution of a rabbit anti-goat alkaline phosphatase labeled secondary antibody (KPL). BCIP-NBT (KPL) was used for colorimetric detection.
Enzymatic digestion of glycans
To evaluate the presence of N-linked glycosylation of the LdNH36-dg2 recombinant protein, a PNGase F assay (New England Biolabs; Ipswitch, MA) was performed according to manufacturer's instructions. The digestion was analyzed with a 4–20% reduced Tris-glycine gel (Life Technologies) transferred to a nitrocellulose membrane. An anti-LdNH36 colorimetric Western blot was performed as describe above.
High performance liquid chromatography
A Shimadzu (Kyoto, Japan) Prominence ultra-fast liquid chromatograph (UFLC) system was used with a photo diode array (PDA) detector and tandem Phenomenex (Torrance, CA) columns (Yarra 3µ SEC-2000 followed by Yarra 3µ SEC-3000). The 50 µL injections were run with a flow rate of 0.5 mL/min in 30 mM Tris pH 7.5, 150 mM NaCl at room temperature. Data analysis was performed on the 280 nm extracted chromatograph. To determine the molecular weight of the peak, a Bio-rad (Hercules, CA) gel filtration standard was injected to create a curve of the distribution coefficient (Kd) versus the log of MW.
Dynamic light scattering (DLS)
Dynamic light scattering was performed with a Wyatt (Santa Barbara, CA) DynaPro 384-well plate reader at room temperature in the SEC200 elution buffer (30 mM Tris pH 7.5, 150 mM NaCl). Data was obtained by averaging 10 measurements, which were collected in 5 s intervals. Dynamic v7 software was used to estimate the hydrodynamic radius from a cumulant fit of the DLS autocorrelation function, assuming a globular protein.
Structural modeling
The structure of LdNH36-dg2 was predicted via comparative modeling, using semi-automatic Swiss-ModelCitation44 and all visualizations/figures were generated with PyMOL (The PyMOL Molecular Graphics System, Schrodinger, LLC). The published structure of a homolog, L. major nucleoside hydrolase, was used as a template.Citation33 PDBsumCitation34 was utilized to determine the amino acids present at the interface of the tetramer.
Liquid chromatography with tandem mass spectrometry (LC-MSE)
A 4–12% Bis-Tris non-reduced SDS-PAGE gel was performed as described above and stained with Coomassie blue. A sample was excised from the bottom, middle, and top portion of the main 36 kDa band. The 64 kDa band was also excised as a fourth sample. Following extraction and trypsin digestion, the samples were analyzed on a NanoAcquity UPLC system in-line with a Synapt mass spectrometer (Waters Corp; Milford, MA). For glycosylation analysis, a double digestion was also performed with trypsin followed by Endo Glu-C. Liquid chromatography separation was achieved with a 180 µm × 20 mm Symmetry C18 (5 µm) trap column (Waters Corp.) in tandem with a 75 µm × 250 mm BEH130 C18 (1.7 µm) (Waters Corp.) and an acetonitrile gradient with formic acid. Protein identification and quantification was performed with ProteinLynx Global Server (PLGS v3.0; Waters Corp.). The Uniprot P. pastoris v. 2013_11 database (5,074 entries) with the LdNH36-dg2 sequence appended was used as the target database. Relative quantification of the abundance of identified proteins was performed with the “high 3′ method.Citation45
Encapsulation of LdNH36-dg2 and CpG-ODN in PLGA microparticles
LdNH36-dg2 protein for making microparticles was produced using an equivalent but smaller scale process as described above. The protein was encapsulated in microparticles using a water-oil-water double emulsion method with modifications.Citation46 LdNH36-dg2 was pre-concentrated using a 10 kDa MWCO Amicon ultra centrifugal filter tube (Millipore) at 10,000 × g to attain a concentration of 30 mg/mL or greater. The concentration was determined by measuring the 280 nm absorbance with a Nanodrop spectrophotometer (Thermo Fisher Scientific) and multiplying the absorbance by the ratio of the molecular weight (34,000 Da) over the extinction coefficient (23,000 M cm−1). A working solution was prepared by diluting the concentrated protein to 20 mg/mL containing 0.5% poly(vinyl alcohol) (PVA, Mw 31,000–50,000, 98–99% hydrolyzed; Sigma-Aldrich) and 50 mg/mL D-mannitol (Sigma-Aldrich). The working solution (100 µL) was homogenized with 100 mg of PLGA (acid terminated, Mw 7,000–17,000, RESOMER® RG 502H Sigma-Aldrich), which was previously dissolved in 2 mL of chloroform, using an IKA® T25 homogenizer (IKA Works, Inc.; Wilmington, NC) at 20,000 rpm for 1 min. The primary emulsion was poured into 13 mL of 5% PVA solution (in 3X PBS) and homogenized at 24,000 rpm for another 2 min. The resulting emulsion was poured into 137 mL of 5% PVA solution (in 3X PBS) and stirred overnight at room temperature to evaporate the chloroform. The microparticles were collected by centrifugation at 5,000 × g, washed twice using Milli-Q water, re-suspended, and lyophilized. Parallel batches of microparticles were pooled during the washing step.
To prepare CpG-loaded microparticles, CpG (ODN 1826 sequence 5′-tccatgacgttcctgacgtt-3′, with phosphorothioate backbone; Integrated DNA Technologies, Inc.; Coralville, IA) was first ion-paired with the cationic lipid 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP; Avanti® Polar Lipids, Inc.; Alabaster, AL) to create a complex that is soluble in dichloromethane, prior to encapsulation in PLGA microparticles by an oil-in-water single emulsion method.Citation46,47 Briefly, 5 mg of CpG in 0.5 mL of IDTE (pH 8) buffer (Tris-EDTA buffer supplied by Integrated DNA Technologies, Inc.) was mixed with DOTAP (previously dissolved in 0.5 mL of dichloromethane) at a molar ratio of 1:1 (nucleotide phosphate: DOTAP cation), and then methanol (1.05 mL) was added into the mixture, which was pipetted until a single-phase solution was obtained. Next, water (0.5 mL) and dichloromethane (0.5 mL) were added sequentially, resulting in phase separation, and the mixture was vortexed and centrifuged at 2,000 × g for 5 min to recover the organic phase containing CpG ion-paired to DOTAP.
The ion-paired CpG was mixed with 125 mg of PLGA dissolved in 1 mL of chloroform, to give a total oil phase volume of ∼2 mL, and then homogenized with 15 mL of 5% PVA solution (in 3X PBS) at 20,000 rpm for 2 min. The resulting emulsion was poured into 185 mL of 5% PVA solution (in 3X PBS) and stirred overnight at room temperature to evaporate the chloroform. The microparticles were collected by centrifugation at 5,000 × g, washed twice using Milli-Q water, re-suspended, and lyophilized. Empty PLGA microparticles were prepared using a single oil-water emulsion method with similar parameters.
Microparticles were imaged by scanning electron microscopy (SEM, JEOL JSM-6100). Protein encapsulation level was measured by Micro BCA™ Protein Assay (Thermo Fisher Scientific) after microparticles were digested in a mixture of DMSO, SDS, and sodium hydroxide solution.Citation48 To quantify CpG encapsulation, microparticles were digested in the same manner, followed by pH adjustment to neutral pH and measurement of CpG concentration by Quant-iT™ OliGreen® ssDNA Assay (Life Technologies; Grand Island, NY). The loading level (μg/mg) is defined as the mass of LdNH36-dg2 protein (or CpG) divided by the mass of microparticles. The loading efficiency (%) is calculated as the actual loading level divided by the nominal loading level, which is defined as the starting LdNH36-dg2 protein (or CpG) mass divided by the starting PLGA mass.
Animals
BALB/c female mice were obtained from Taconic Farms, Inc. (Hudson, NY). All mice were housed in the Texas Children's Hospital Feigin Center animal facility, operated by the Center for Comparative Medicine at Baylor College of Medicine (BCM). Experiments were conducted under an animal research protocol approved by the Institutional Animal Care and Use Committee at BCM and followed the BCM Guidelines for Animal Care and Use.
Vaccinations and sample collection
Lyophilized microparticles were re-suspended in sterile PBS and vortexed for approximately 10 s prior to mixing the vaccines. Female BALB/c mice (n = 4 per group) were vaccinated with 40 µg of LdNH36-dg2 plus varying amounts of CpG adjuvant (10, 20, 40, or 80 μg), encapsulated in separate PLGA microparticles. Vaccine control groups included microparticle-LdNH36-dg2 and soluble LdNH36-dg2 + CpG. Negative control groups included microparticle-CpG, empty microparticles, and PBS. Mice were vaccinated in 2 cohorts of 5 groups each, separated by 2 d. As an internal control, the MP/LdNH36-dg2/CpG group with 20 μg CpG was included in both cohorts. After 3 weeks, a boost vaccination was given on the contralateral side. In each vaccination, mice were given a single injection of 100 μL of the designated vaccine subcutaneously in the flank using a 1 mL TB syringe 25G × 5/8 (BD; Franklin Lakes, NJ).
Two weeks after the boost, mice were euthanized by exsanguination via cardiac puncture under the anaesthetized condition by intraperitoneal injection of a dose of 3 mg ketamine (Ketaved™, distributed by VEDCO Inc.; St Joseph, MO) and 0.3 mg xylazine (AnaSed™, Akorn, Inc.; Decatur, IL). Cervical dislocation was performed to ensure death. Blood samples were allowed to coagulate in Serum-Gel clotting tubes (SARSTEDT, Inc.) at room temperature for 2 h and centrifuged at 10,000 × g for 5 min to separate serum. Serum samples were stored at −20°C before performing serum antibody ELISAs.
Serum antibody enzyme linked immunosorbent assay (ELISA)
Antigen-specific serum antibody binding was measured by ELISA. Briefly, 96-well plates were sensitized overnight at 4˚C with 100 ng/well of antigen (LdNH36-dg2 protein) in 50 μL of PBS. Plates were aspirated and blocked with 100 μL PBS plus 2% bovine serum albumin (BSA; KPL Inc; Gaithersburg, MD) for 2 h at room temperature, and then incubated for 2 h with 50 μL of serum samples that were serially diluted at 1:20 to 1:9.77 × 108 in PBST containing 0.1% BSA. After washing with PBST, plates were incubated for 1 h with 50 μL of a 1:4,000 dilution of horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG1, IgG2a, and IgG2b antibodies (LifeSpan Biosciences, Inc.; Seattle, WA). Plates were washed 6 times, and HRP was quantified by adding 100 μL of TMB Substrate (Thermo Fisher Scientific; Rockford, IL). After 8–10 min, the reaction was stopped with 100 μL of 1 M HCl, and the optical density (OD) was measured at 450 nm using a SpectraMax® M3 microplate reader (Molecular Devices, LLC; Sunnyvale, CA). Titers were determined by a log-linear interpolation of the serum dilution value at the cut-off OD, which was determined by the average OD of 1:1,600 diluted serum from naïve mice plus 3 times the standard deviation.Citation49 In order to determine the reactivity of the serum antibodies to wild-type LdNH36, antibody titers were measured using LdNH36-E-WT (non-polyhistidine tagged) as the plate-coating protein. The same procedure described above was followed, except the sera had been stored at −20°C for 2 y.
Statistical analysis
Antibody titers were log-transformed. The effect of vaccine formulations to antibody titers in response to either LdNH36-E-WT or LdNH36-dg2 was analyzed by one-way ANOVA followed by a Tukey multiple comparison test, and the differences of antibody titers to LdNH36-E-WT and LdNH36-dg2 were compared using pairwise t test. The analysis was performed using either R software (RStudio; Boston, MA) or SAS (SAS Institute; Cary, NC) with p value of 0.05. Data from negative control groups (microparticle-CpG, empty microparticle, and PBS) were excluded from data analysis (titers < 103). All data plotting was conducted using Prism software (GraphPad Software, Inc.).
Antibody inhibition assay
Nucleoside hydrolase activity was assayed at room temperature in 50 mM Tris-HCl pH 7.5. Using a 96 well format and Epoch microplate spectrophotometer (BioTek; Winooski, VT), the hydrolysis of inosine to hypoxanthine and ribose was determined by measuring absorbance changes at 280 nm.50 An inosine standard curve was used to convert absorbance units to concentration units (1197 AU = 1000 µM). Total IgG was purified from mouse sera with Pierce Protein G UltraLink Resin (Thermo Fisher Scientific) and concentrated with Millipore Amicon 0.5 mL spin cartridges (10 kDa MWCO).
Disclosure of potential conflicts of interest
Several authors of this manuscript currently are involved in a program for the development of a leishmaniasis vaccine. There are no other conflicts of interest to disclose.
HVI_LdNH36-dg2_Supplemental_Figures_1-3.zip
Download Zip (333.4 KB)Acknowledgments
We thank our collaborators, Dr. Jesus G. Valenzuela and Dr. Shaden Kamhawi (National Institute of Allergy and Infectious Disease, Vector Molecular Biology Section, Laboratory of Malaria and Vector Research), for helpful discussions throughout this project. We are grateful to Dr. Kathryn Jones (BCM) for providing the anti-LdNH36 mouse polyclonal sera. Also, we would like to thank the Department of Pathology at Texas Children's Hospital for the use of their Electron Microscopy Core Facility.
References
- GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015; 385:117-71; PMID:25530442; http://dx.doi.org/10.1016/S0140-6736(14)61682-2
- Hotez PJ. Forgotten People, Forgotten Diseases: The Neglected Tropical Diseases and Their Impact on Global Health and Development ASM Press, 2013
- Lee BY, Bacon KM, Shah M, Kitchen SB, Connor DL, Slayton RB. The economic value of a visceral leishmaniasis vaccine in Bihar State, India. Am J Tropical Med Hyg 2012; 86:417-25; http://dx.doi.org/10.4269/ajtmh.2012.10-0415
- Bacon KM, Hotez PJ, Kruchten SD, Kamhawi S, Elena M, Valenzuela JG, Lee BY. The potential economic value of a cutaneous leishmaniasis vaccine in seven endemic countries in the Americas. Vaccine 2013; 31:480-6; PMID:23176979; http://dx.doi.org/10.1016/j.vaccine.2012.11.032
- Aguilar-be I, Zardo S, Souza EPD, Patrı G, Rosado-vallado M, Mut-martin M, Rosario M, Beatriz C, Sousa PD, Dumonteil E. Cross-Protective Efficacy of a Prophylactic Leishmania donovani DNA Vaccine against Visceral and Cutaneous Murine Leishmaniasis. Infection Immunity 2005; 73:812-9; PMID:15664920; http://dx.doi.org/10.1128/IAI.73.2.812-819.2005
- Chale-balboa W, Mut-Martin M, Ramirez-Sierra M, Garcia-Miss M, Dumonteil E. A combination DNA vaccine encoding nucleoside hydrolase 36 and glycoproteine 63 protects female but not male hamsters against Leishmania mexicana. Parasite 2009; 16:227-30; PMID:19839269; http://dx.doi.org/10.1051/parasite/2009163227
- Boitz JM, Ullman B, Jardim A, Carter N. Purine salvage in Leishmania : complex or simple by design? Trends Parasitol 2013; 28:345-52; http://dx.doi.org/10.1016/j.pt.2012.05.005
- Datta AK, Datta R, Sen B. Antiparasitic chemotherapy: Tinkering with the purine salvage pathway. Adv Exp Med Biol 2008; 625:116-32; PMID:18365663; http://dx.doi.org/10.1007/978-0-387-77570-8_10
- Cui L, Rajasekariah GR, Martin SK. A nonspecific nucleoside hydrolase from Leishmania donovani: implications for purine salvage by the parasite. Gene 2001; 280:153-62; PMID:11738828; http://dx.doi.org/10.1016/S0378-1119(01)00768-5
- Palatnik-de-Sousa CB, Dutra HS, Borojevic R. Leishmania donovani surface glycoconjugate GP36 is the major immunogen component of the Fucose-Mannose Ligand (FML). Acta Tropica 1993; 53:59-72; PMID:8096110; http://dx.doi.org/10.1016/0001-706X(93)90006-W
- Palatnik de Sousa C, Gomes E, de Souza E, dos Santos W, de Macedo S, de Medeiros L, Luz K. The FML (fucose mannose ligand) of Leishmania donovani: a new tool in diagnosis, prognosis, transfusional control and vaccination against human kala-azar. Rev Soc Bras Med Trop 1996; 29:153-63; PMID:8713607; http://dx.doi.org/10.1590/S0037-86821996000200008
- Gamboa-León R, Paraguai de Souza E, Borja-Cabrera GP, Santos FN, Myashiro LM, Pinheiro RO, Dumonteil E, Palatnik-de-Sousa CB. Immunotherapy against visceral leishmaniasis with the nucleoside hydrolase-DNA vaccine of Leishmania donovani. Vaccine 2006; 24:4863-73; http://dx.doi.org/10.1016/j.vaccine.2006.03.005
- Li L, Saade F, Petrovsky N. The future of human DNA vaccines. J Biotechnol 2013; 162:171-82; http://dx.doi.org/10.1016/j.jbiotec.2012.08.012
- Grunwald T, Ulbert S. Improvement of DNA vaccination by adjuvants and sophisticated delivery devices : vaccine- platforms for the battle against infectious diseases. Clin Exp Vacc Res 2015; 4:1-10; http://dx.doi.org/10.7774/cevr.2015.4.1.1
- Al-wabel MA, Tonui WK, Cui L, Martin SK, Titus RG. Protection of Susceptible BALB / c Mice from Challenge with Leishmania major by Nucleoside Hydrolase, a Soluble Exo-antigen of Leishmania. Am J Tropical Med Hyg 2007; 77:1060-5
- Ferrer-Miralles N, Domingo-Espín J, Corchero JL, Vázquez E, Villaverde A. Microbial factories for recombinant pharmaceuticals. Microbial Cell Factories 2009; 8:17; PMID:19317892; http://dx.doi.org/10.1186/1475-2859-8-17
- Buckholz R, Gleeson M. Yeast Systems for the Commercial Production of Heterologous Proteins. Nat Biotechnol 1991; 9:1067-72; http://dx.doi.org/10.1038/nbt1191-1067
- Cregg JM, Vedvick T, Raschke W. Recent Advances in the Expression of Foreign Genes in Pichia pastoris. Nat Biotechnol 1993; 11:905-10; http://dx.doi.org/10.1038/nbt0893-905
- Rabert C, Weinacker D, Pessoa A, Jr, Farias J. Recombinants proteins for industrial uses : utilization of Pichia pastoris expression system. Brazilian J Microbiol 2013; 44:351-6; http://dx.doi.org/10.1590/S1517-83822013005000041
- Gemmill TR, Trimble RB. Overview of N - and O -linked oligosaccharide structures found in various yeast species. 1999; 1426:227-37; PMID:9878752
- Kocken CHM, Withers-Martinez C, Dubbeld Ma, van der Wel A, Hackett F, Blackman MJ, Thomas AW. High-Level Expression of the Malaria Blood-Stage Vaccine Candidate Plasmodium falciparum Apical Membrane Antigen 1 and Induction of Antibodies That Inhibit Erythrocyte Invasion. Infection Immunity 2002; 70:4471-6; PMID:12117958; http://dx.doi.org/10.1128/IAI.70.8.4471-4476.2002
- Liu M, Chen H, Luo F, Li P, Pan Q, Xia B, Qi Z, Ho WZ, Zhang XL. Deletion of N-glycosylation sites of hepatitis C virus envelope protein E1 enhances specific cellular and humoral immune responses. Vaccine 2007; 25:6572-80; PMID:17675185; http://dx.doi.org/10.1016/j.vaccine.2007.07.003
- Mori K, Sugimoto C, Ohgimoto S, Nakayama EE, Shioda T, Kusagawa S, Takebe Y, Kano M, Matano T, Yuasa T, et al. Influence of glycosylation on the efficacy of an Env-based vaccine against simian immunodeficiency virus SIVmac239 in a macaque AIDS model. J Virol 2005; 79:10386-96; PMID:16051831; http://dx.doi.org/10.1128/JVI.79.16.10386-10396.2005
- Chen WH, Du L, Chag SM, Ma C, Tricoche N, Tao X, Seid CA, Hudspeth EM,Lustigman S, Tseng CTK, et al. Yeast-expressed recombinant protein of the receptor-binding domain in SARS-CoV spike protein with deglycosylated forms as a SARS vaccine candidate. Hum Vacc Immunotherapeutics 2014; 10:1-11; http://dx.doi.org/10.4161/hv.28050
- Kaye P, Scott P. Leishmaniasis: complexity at the host-pathogen interface. Nat Rev Microbiol 2011; 9:604-15; PMID:21747391; http://dx.doi.org/10.1038/nrmicro2608
- Johansen P, Men Y, Merkle HP, Gander B. Revisiting PLA/PLGA microspheres: An analysis of their potential in parenteral vaccination. Eur J Pharmaceutics Biopharmaceutics 2000; 50:129-46; http://dx.doi.org/10.1016/S0939-6411(00)00079-5
- O'Hagan DT, Singh M, Gupta RK. Poly(lactide-co-glycolide) microparticles for the development of single-dose controlled-release vaccines. Adv Drug Delivery Rev 1998; 32:225-46; http://dx.doi.org/10.1016/S0169-409X(98)00012-X
- Oyewumi M, Kumar A, Cui Z. Nano-microparticles as immune adjuvants: correlating particle sizes and the resultant immune response. Expert Rev Vacc 2010; 9:1095-107; http://dx.doi.org/10.1586/erv.10.89
- Klinman DM, Klaschik S, Sato T, Tross D. CpG oligonucleotides as adjuvants for vaccines targeting infectious diseases. Adv Drug Delivery Rev 2009; 61:248-55; http://dx.doi.org/10.1016/j.addr.2008.12.012
- Jaafari MR, Badiee A, Khamesipour A, Samiei A, Soroush D, Kheiri MT, Barkhordari F, McMaster WR, Mahboudi F. The role of CpG ODN in enhancement of immune response and protection in BALB/c mice immunized with recombinant major surface glycoprotein of Leishmania (rgp63) encapsulated in cationic liposome. Vaccine 2007; 25:6107-17; PMID:17629372; http://dx.doi.org/10.1016/j.vaccine.2007.05.009
- Rhee EG, Mendez S, Shah JA, Wu CY, Kirman JR, Turon TN, Davey DF, Davis H, Klinman DM, Coler RN, et al. Vaccination with heat-killed leishmania antigen or recombinant leishmanial protein and CpG oligodeoxynucleotides induces long-term memory CD4+ and CD8+ T cell responses and protection against leishmania major infection. J Exp Med 2002; 195:1565-73; PMID:12070284; http://dx.doi.org/10.1084/jem.20020147
- Ebrahimpoor S, Pakzad SR, Ajdary S. IgG1 and IgG2a Profile of Serum Antibodies to Leishmania major Amastigote in BALB / c and C57BL / 6 Mice. 2013; 12:361-7
- Shi W, Schramm V, Almo S. Nucleoside Hydrolase from Leishmania major. cloning, expression, catalytic properties, transition state inhibitors, and the 2.5-a crystal structure. J Biol Chem 1999; 274:21114-20; PMID:10409664; http://dx.doi.org/10.1074/jbc.274.30.21114
- De Beer TaP, Berka K, Thornton JM, Laskowski Ra. PDBsum additions. Nucleic Acids Res 2014; 42:292-6; http://dx.doi.org/10.1093/nar/gkt940
- Khurshid S, Saridakis E, Govada L, Chayen NE. Porous nucleating agents for protein crystallization. Nat Protocols 2014; 9:1621-33; PMID:24922271; http://dx.doi.org/10.1038/nprot.2014.109
- Garrity R, Rimmelzwaan G, Minassian A, Tsai W, Lin G, de Jong J, Goudsmit J, Nara P. Refocusing Neutralizing Antibody Response. J Immunol 1997; 159:279-89; PMID:9200464
- Nico D, Claser C, Borja-Cabrera GP, Travassos LR, Palatnik M, Soares IDS, Rodrigues MM, Palatnik-de-Sousa CB. Adaptive immunity against Leishmania nucleoside hydrolase maps its c-terminal domain as the target of the CD4+ T cell-driven protective response. PLoS Neglected Tropical Dis 2010; 4:e866; http://dx.doi.org/10.1371/journal.pntd.0000866
- Paraguai de Souza E, Bernardo R, Palatnik M, Sousa C. Vaccination of Balb / c mice against experimental visceral leishmaniasis with the GP36 glycoprotein antigen of Leishmania donovani. Vaccine 2001; 19:3104-15; PMID:11312005; http://dx.doi.org/10.1016/S0264-410X(01)00031-7
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein Identification and analysis tools on the ExPASy Server. Methods Mol Biol 2005; 112:571-608
- Liu C, Hashizume T, Kurita-Ochiai T, Fujihashi K, Yamamoto M. Oral immunization with Porphyromonas gingivalis outer membrane protein and CpGoligodeoxynucleotides elicits T helper 1 and 2 cytokines for enhanced protective immunity. Mol Oral Microbiol 2010; 25:178-89; PMID:20502628; http://dx.doi.org/10.1111/j.2041-1014.2009.00560.x
- Chu RS, Targoni OS, Krieg aM, Lehmann PV, Harding CV. CpG oligodeoxynucleotides Act as Adjuvants that Switch on T Helper 1 (Th1) Immunity. J Exp Med 1997; 186:1623-31; PMID:9362523; http://dx.doi.org/10.1084/jem.186.10.1623
- Gupta R, Jung E, Brunak S. Predication of N-glycosylation sites in human proteins. Center for Biological Sequence Analysis at Technical University of Denmark DTU, 2004
- Stratton J, Chiruvolu V, Meagher M. High cell-density fermentation. Meth Mol Biol (Clifton, NJ) 1998; 103:107-20
- Guex N, Peitsch MC, Schwede T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 2009; 30 Suppl 1:S162-73; PMID:19517507; http://dx.doi.org/10.1002/elps.200900140
- Silva JC, Gorenstein MV, Li GZ, Vissers JPC, Geromanos SJ. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol Cell Proteomics 2006; 5:144-56; PMID:16219938; http://dx.doi.org/10.1074/mcp.M500230-MCP200
- Wang Q, Tan MT, Keegan BP, Barry MA, Heffernan MJ. Time course study of the antigen-specific immune response to a PLGA microparticle vaccine formulation. Biomaterials 2014; 35:8385-93; PMID:24986256; http://dx.doi.org/10.1016/j.biomaterials.2014.05.067
- Patel MM, Zeles MG, Manning MC, Randolph TW, Anchordoquy TJ. Degradation kinetics of high molecular weight poly(L-lactide) microspheres and release mechanism of lipid:DNA complexes. J Pharmaceutical Sci 2004; 93:2573-84; http://dx.doi.org/10.1002/jps.20176
- Sah H. A new strategy to determine the actual protein content of poly(lactide-co-glycolide) microspheres. J Pharmaceutical Sci 1997; 86:1315-8; http://dx.doi.org/10.1021/js960363q
- Frey A, Di Canzio J, Zurakowski D. A statistically defined endpoint titer determination method for immunoassays. J Immunological Meth 1998; 221:35-41; http://dx.doi.org/10.1016/S0022-1759(98)00170-7
- Parkin DW, Horensteingn Ba, Abdulahg DR, Estupiiang B, Schrammgii VL. Nucleoside Hydrolase from Crithidia fasciculuta. J Biol Chem 1991; 266:20658-65; PMID:1939115