
ABSTRACT
Background: The influenza B virus has two lineages; Yamagata and Victoria. The two lineages are antigenically distinct and it is difficult to expect cross-protection between the lineages. Actually, the mismatch between circulating influenza B viruses and vaccine strains has been occurred frequently. The cell-culture system for the production of influenza vaccine can contribute to improve vaccine strain selection and expand vaccine supplies. We investigated the immunogenicity and safety of cell culture-derived quadrivalent inactivated influenza vaccine (NBP607-QIV) in adults and elderly subjects.
Methods: A randomized controlled phase III trial was undertaken in 10 university hospitals in the Republic of Korea (Clinical trial Number—NCT02467842). Adults (aged 19–59 years) and elderly subjects (aged ≥60 years) were randomly assigned in a 2:1:1 ratio to NBP607-QIV versus cell culture-based trivalent inactivated influenza vaccine-Yamagata (NBP607-Y) and cell culture-based trivalent inactivated influenza vaccine-Victoria (NBP607-V). Immunogenicity was assessed 3 weeks after vaccination by hemagglutination inhibition assay. Safety was assessed for 6 months post-vaccination: solicited adverse events (AEs) for 7 days, unsolicited AEs for 21 days and serious adverse events (SAEs) for 6 months. AEs were sub-classified as adverse drug reactions (ADRs) according to the causality.
Results: A total of 1,503 participants were randomly assigned to NBP607-QIV (n = 752), NBP607-Y (n = 373) and NBP607-V (n = 378). The seroconversion rates of NBP607-QIV were 52.4%, 51.2%, 43.7% and 55.8% against A/H1N1, A/H3N2, B/Yamagata and B/Victoria, respectively. Non-inferiority against shared strains and superiority against alternate-lineage B strains were demonstrated for NBP607-QIV vs. NBP607-Y and NBP607-V. A total of 730 reactions occurred in 324 (43.1%) subjects of NBP607-QIV group. Majority of ADRs was solicited (99.2%) and mild (90.3%) in intensity. In adults (aged 19–59 years), solicited local AEs were slightly more frequent in NBP607-QIV group than NBP607-Y or NBP607-V group (40.9%, 33.4% and 32.5%, respectively). One SAE was observed among NBP607-QIV group, which was considered to be unrelated to the study vaccine within 3 weeks of vaccination and no vaccine-related SAEs were reported up to 6 months after vaccination.
Conclusions: NBP607-QIV is a safe, well-tolerated and immunogenic influenza vaccine in Korean adults and elderly subjects.
Introduction
For years, influenza vaccines were designed to protect against three different flu viruses, which included an influenza A/H1N1 virus, an influenza A/H3N2 virus and one B virus. However, influenza B strains have diverged into two lineages, known as Victoria and Yamagata.Citation1 The two lineages are antigenically distinct and it is difficult to expect cross-protection between the lineages.Citation2 Actually, the mismatch between circulating influenza B viruses and vaccine strains has been occurred frequently and the B lineage selected for seasonal vaccines has matched the dominant circulating strain only about half the time.Citation3,4 To address this mismatch, the WHO has recommended a fourth strain to be included in seasonal influenza vaccines since 2012.Citation5 The cell-culture influenza vaccines are being developed as an alternative to the egg-based manufacturing process because the technology is more flexible than the traditional technology, which relies upon adequate supply of eggs.Citation6 The main benefit of the cell-culture system is the ability to rapidly produce vaccine supplies during an impending pandemic and the avoidance of egg-related allergic reactions. SK Chemicals has developed cell culture-derived quadrivalent inactivated influenza vaccine (NBP607-QIV, Andong-si, SK Chemicals, Republic of Korea) first in the world. We investigated the immunogenicity and safety of the NBP607-QIV in adults and elderly subjects.
Results
Study subjects
A total of 1,514 subjects were screened and 1,503 were enrolled to be randomized between October and November, 2014. The enrolled subjects were randomized to the NBP607-QIV (752 subjects), culture-based trivalent inactivated influenza vaccine containing B/Yamagata (NBP607-Y, 373 subjects), or culture-based trivalent inactivated influenza vaccine containing B/Victoria (NBP607-V, 378 subjects) group (). Baseline characteristics were well matched between the three groups, as shown in . All the subjects were included in the safety analysis. A total of 8 subjects were excluded from the per-protocol analysis because of the protocol deviation.
Figure 1. Flow chart of the subjects throughout the study.
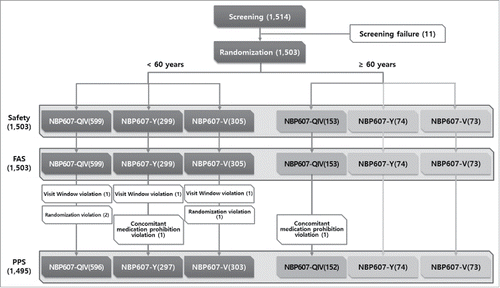
Table 1. Baseline characteristics of the study subjects.Footnotea
Immunogenicity
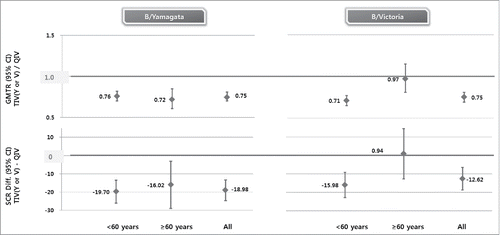
NBP607-QIV met non-inferiority criteria against shared strains and superiority criteria against alternate-lineage B strains vs. cell culture-based trivalent inactivated influenza vaccine (NBP607-TIV) (, ). As for the shared strains, the geometric mean titer ratio (GMTR) of NBP607-TIV/NBP607-QIV was 1.02 (95% CI, 0.94 to 1.10) for A/H1N1 stain, 0.94 (95% CI, 0.87 to 1.01) for A/H3N2 strain, 0.88 (95% CI, 0.82 to 0.95) for B/Yamagata strain, and 0.90 (95% CI, 0.83 to 0.97) for B/Victoria strain. The seroconversion rate (SCR) difference (NBP607-TIV minus NBP607-QIV) was −1.00 (95% CI, −6.07 to 4.06) for A/H1N1, −5.02 (95% CI, −10.08 to 0.04) for A/H3N2, −7.06 (95% CI, −13.12 to −1.00) for B/Yamagata strain, and −3.09 (95% CI, −9.26 to 3.09) for B/Victoria strain. As for the alternate-lineage B strains, the GMTR was 0.75 (95% CI, 0.70 to 0.81) for B/Yamagata and 0.75 (95% CI, 0.69 to 0.81) for B/Victoria. The SCR difference was −18.98 (95% CI, −24.61 to −13.36) for B/Yamagata and −12.62 (95% CI, −18.79 to −6.45) for B/Victoria.
Figure 2. Non-inferiority of NBP607-QIV over NBP607-TIV on immunogenicity for each strain at 21 days post-vaccination. The horizontal bold line indicates non-inferiority threshold. According to the CBER guidance criterion for noninferiority, for each of the 4 strains 1) the upper limit of the 2-sided 95% CI on the difference between the seroconversion rates (TIV−QIV) must be <10%; 2) the upper limit of the 2-sided 95% CI for the ratio of GMTs (GMT TIV/GMT QIV) for HI antibody should be <1.5.
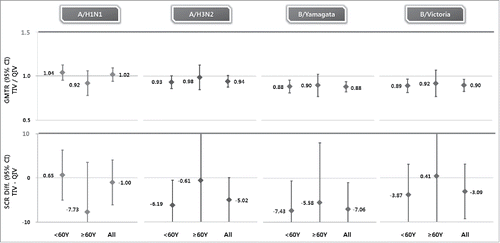
Figure 3. Superiority of NBP607-QIV over NBP607-TIV on immunogenicity for B/Yamagata or B/Victoria strain at 21 days post-vaccination. The horizontal bold line indicates superiority threshold. For superiority, 1) the upper limit of the 2-sided 95% CI on the difference between the seroconversion rates (TIV−QIV) must be <0%; 2) the upper limit of the 2-sided 95% CI for the ratio of GMTs (GMT TIV/GMT QIV) for HI antibody should be <1.0.
NBP607-QIV met the Committee for Medicinal Products for Human Use (CHMP) criteria in young adults (19–59 years) and the elderly (≥60 years) (). Seroprotection rates were 98.3% for A/H1N1, 99.5% for A/H3N2, 98.5% for B/Yamagata and 99.2% for B/Victoria in subjects aged 19–59 years, and were 92.8% for A/H1N1, 98.7% for A/H3N2, 94.1% for B/Yamagata and 96.1% for B/Victoria in subjects aged ≥60 years. The seroconversion rates were 52.4% for A/H1N1, 53.5% for A/H3N2, 43.8% for B/Yamagata and 54.7% for B/Victoria in subjects aged 19–59 years, and were 52.6% for A/H1N1, 42.1% for A/H3N2, 43.4% for B/Yamagata and 59.9% for B/Victoria in subjects aged ≥60 years. The GMTRs ranged from 3.2 to 4.8 in subjects aged 19–59 years and from 3.0 to 4.6 in subjects aged ≥60 years.
Table 2. Immunogenicity at 21-day after vaccination.
Safety
A total of 860 adverse events (AEs) occurred in 365 (48.5%) of 752 subjects in NBP607-QIV group, while 385 events in 169 (45.3%) of 373 subjects in NBP607-Y group and 372 events in 159 (42.1%) of 378 subjects. In terms of the adverse drug reaction (ADR), 730 reactions occurred in 324 (43.1%) subjects of NBP607-QIV group, 324 reactions in 144 (38.6%) subjects of NBP607-Y group and 325 reactions in 146 (38.6%) subjects of NBP-V group. Majority of ADRs was solicited local (99.2%) and mild (90.3%) in intensity. The solicited local and systemic ADRs reported within 7 days of vaccination are shown in . The solicited local ADRs were more frequent in NBP607-QIV group than NBP607-TIV group (p = 0.04). However, there was no significant difference in the incidence of solicited systemic ADRs between NBP607-QIV group and NBP607-TIV group. A total of 6 unsolicited ADRs were reported in NBP607-QIV group, 2 reactions in NBP607-Y group and 8 reactions in NBP607-V group. Unsolicited reactions include chills, injection site pruritus, injection site warmth, injection site pain, abdominal pain, gastroesophageal reflux disease, nausea, esophagitis, musculoskeletal pain, flushing, nasopharyngitis and eczema. One severe adverse event (SAE) was observed among NBP607-QIV group within 3 weeks of vaccination. It was nasal septum deviation and was considered to be unrelated to the study vaccine. And no vaccine-related SAEs were reported up to 6 months after vaccination.
Table 3. Solicited local and systemic adverse drug reactions within 7 days after vaccination.
Discussion
NBP607-QIV is a MDCK cell culture-derived inactivated, subunit, quadrivalent influenza vaccine, which is the first in the world. With the limitation of egg-based influenza vaccine production system, there has been a need for the cell culture-derived production system. In particular, prompt and large production of vaccine is one of the most important requirements for better respond to the influenza pandemic.Citation7 Actually, during the 2009 A/H1N1pdm influenza pandemic, vaccine supply was started too late.Citation8,9 In order to respond properly to the influenza pandemic, it is preferable that cell culture-derived influenza vaccine production system is operated inter-pandemic phase. The SK Chemicals already developed cell culture-derived trivalent influenza vaccine (NBP607) and the vaccine was started to be used in Korea from 2015–2016 influenza season.Citation10 So, this clinical trial was conducted using NBP607 as a control vaccine.
To address for the need for quadrivalent seasonal influenza vaccine, six kinds of quadrivalent seasonal influenza vaccines were developed and licensed to date: Flumist Quadrivalent of MedImmune, Fluarix Quadrivalent (QIV-008) and FluLaval Quadrivalent of GlaxoSmithKline, Fluzone Quadrivalent (GRC43) and Fluzone Intradermal Quadrivalent of Sanofi Pasteur, and Flucelvax of Seqirus Inc.Citation11-15 Comparing with the clinical trials results of those vaccines, NBP607-QIV showed comparable results in immunogenicity and safety. With the results, Korea Ministry of Food and Drug Safety (MFDS) authorized the use of NBP607-QIV in December 2015. The Flucelvax is another cell culture-derived quadrivalent influenza vaccine, which was licensed in May 2016 in United States. So, NBP607-QIV is the 6th quadrivalent seasonal influenza vaccine, and the first licensed cell culture-derived quadrivalent inactivated influenza vaccine in the world.
One of the control vaccines, NBP607-Y, was shown to have partial effective immune response for the B/Victoria. In a previous study performed with quadrivalent influenza vaccine, similar immune response was observed with the trivalent vaccine containing B/Yamagata lineage only.Citation16 The study performed with B/Yamagata monovalent vaccine also reported similar results.Citation17 However, the immune response for the B/Victoria induced by NBP607-Y was lower than those induced by NBP607-QIV or NBP607-V. It means that the trivalent vaccine containing B/Yamagata lineage cannot be substituted for quadrivalent vaccine.
Adverse events, specially pain or tenderness, were observed more often in NBP607-QIV group. It is thought to occur because NBP607-QIV contained more antigens. However, the incidence of solicited local ADR was not so high compared to the previous study results, although the head-to-head comparison is not appropriate. And the most of the ADR was mild in severity and showed self-limiting progress without specific treatment.
This study has a few limitations. First, only HI assay was used as an immunologic assessment. However, HI assay is the standard immunologic assessment for an influenza vaccine, and the influenza vaccine authorizing criteria of CHMP and MFDS is based on the HI assay. Second, the immunologic assay was performed only for 3–4 weeks after vaccination. Considering previous study results performed with NBP607-TIV, however, it can be assumed that the immunity induced by NBP607-QIV will be maintained as sufficient level for 6 months.
In conclusion, NBP607-QIV, the first cell culture-derived quadrivalent inactivated influenza vaccine, is a safe, well-tolerated and immunogenic influenza vaccine in Korean adults and elderly subjects.
Materials and methods
Study design
A multi-center, randomized, double-blind phase III clinical trial was undertaken to assess the immunogenicity and safety of NBP607-QIV in adults and elderly subjects at 10 university hospitals of the Republic of Korea (ROK) during 2014–2015 season (Clinical trial Number—NCT02467842). The first subject recruitment was on October 10, 2014, and the second blood sampling of the lase subjects was done on December 10, 2014.
Healthy adults aged 19 years or older were enrolled. Subjects were excluded for the following reasons: disorders in immune function, any malignancy or lymphoproliferative disorder, history of Guillain-Barre syndrome, known bleeding diathesis or any condition that may be associated with a prolonged bleeding time, experience of fever (>38.0°C) within 24 hours prior to vaccination, body temperature >38.0°C at the vaccination day, concomitant medications/therapy such as immunosuppressants or immune modifying drugs, systemic corticosteroids, immunoglobulins, blood or blood- derived products within 3 months, influenza vaccination within 6 months, administration of other investigation products within 4 weeks, any vaccination within 1 month, planning to receive any vaccine within 1 month from the study vaccine, any serious chronic or progressive disease, pregnant or breast-feeding women. Immunogenicity was assessed 3 weeks after vaccination. Safety was assessed for 6 months post-vaccination: solicited AEs for 7 days, unsolicited AEs for 21 days, and SAEs for 6 months.
At visit 1, written informed consents were obtained from all the subjects. After screening process, subjects were randomly assigned in a 2:1:1 ratio to NBP607-QIV vs. two kinds of cell culture-based trivalent inactivated influenza vaccine, NBP607-Y and NBP607-V. The participants were randomly assigned to each study group using the block randomization method and treatment allocation was performed using the interactive web-based response system. Baseline blood samples were collected just before the vaccination. The participants received a single dose of their assigned vaccine intramuscularly into the deltoid muscle. They were observed for 30 minutes after vaccination. On days 3–7 (visit 2), a telephone interview was conducted to assess the short-term safety. A second blood sample was obtained on days 21–28 after vaccination (visit 3). On days 150–210 (visit 4), a telephone interview was conducted to assess the long-term safety.
The vaccines for clinical trial were in the form of a pre-filled syringe of the same shape, so that the subjects could not distinguish the vaccine type. For maintaining double-blind, a non-blind pharmacist was designated for each institution. Each institution's non-blind pharmacist was obliged to maintain a qualitative level of blindness for administration of the drug for clinical trials. In addition, non-blindness monitors were responsible for checking the quantity and dispensing of investigational product at each testing laboratory. Non-blind pharmacists and non-blind monitors should only perform pre-designated tasks related to clinical trials, and do not engage in other tasks.
The primary outcome measures were the non-inferiority of NBP607-QIV over NBP607-TIV on immunogenicity for each strain at day 21 post vaccination in all subjects and meeting the CHMP criteria in the elderly subjects aged ≥65 years. The secondary outcome measures were the superiority of NBP607-QIV over NBP607-TIV on immunogenicity for the alternate-lineage B strain in all subjects, CHMP criteria in the adults aged 19–59 years. The safety assessment included numbers of participants with AEs, and numbers of participants with SAE.
Vaccines
The study vaccine (NBP607-QIV) was a MDCK cell culture-derived quadrivalent inactivated subunit influenza vaccine containing 15 µg of hemagglutinin per strain. The vaccine was prepared by SK Chemicals (Andong-si, ROK) in the form of a 0.5 mL prefilled syringe. According to the recommendations of the WHO, it included the following four strains: A/California/7/2009 (H1N1)pdm09-like virus, A/Texas/50/2012 (H3N2)-like virus, B/Massachusetts/2/2012-like virus and B/Brisbane/60/2008-like virus. The two types of control vaccines were MDCK cell culture-derived trivalent inactivated subunit influenza vaccines containing 15 µg of hemagglutinin per strain. The vaccines were also prepared by SK Chemicals in the same form of a 0.5 mL prefilled syringe. The control vaccines contained only one among the two lineages of influenza B virus strains.
Immunogenicity assessment
Immunogenicity was assessed by hemagglutination inhibition (HI) assay. The HI antibodies to each hemagglutinin of the A/H1N1, A/H3N2, B/Yamagata and B/Victoria strains contained in the vaccine using a standard assay using the cell-derived HA antigens and chicken erythrocytes. The HI assay was performed in the SK Chemicals Life Science Research Institute.Citation10 HI antibody titers that were below the detection limit (<1:10) were assigned a value of 1:5.
The non-inferiority of adjusted GMT (adjusted for the baseline titer) and SCR was assessed at Week 3 for NBP607-QIV vs. NBP607-Y+NBP607-V against all four strains. And the superiority of adjusted GMT (adjusted for the baseline titer) and SCR was assessed at Week 3 for NBP607-QIV vs. NBP607-V against the B/Yamagata strain and NBP607-QIV vs. NBP607-Y against the B/Victoria strain.
The three immunogenicity end points after vaccination were the proportion of subjects with antibody titers of 1:40 or more on HI assay (seroprotection rate), the proportion of subjects with a change in HI titer from <1:10 to ≥1:40, or a 4-fold or more increase in antibody titer (seroconversion rate), and GMTR [the ratio of the geometric mean titer (GMT) after vaccination to GMT before vaccination]. Serologic response was assessed using the CHMP criteria.Citation18 In order to confirm protective immunogenicity based on the CHMP criteria, all of the following criteria must be met: seroprotection rates >70% for subjects aged 19–59 years and >60% for subjects aged ≥60 years, seroconversion rates>40% for subjects aged 19–59 years and >30% for subjects aged ≥60 years, or GMTR >2.5 for subjects aged 19–59 years and >2.0 for subjects aged ≥60 years.
Safety assessment
At the first visit, enrolled subjects were given a thermometer, a diary card for recording AEs and a numerical scale. The subjects were asked to record the lists and severity of AEs, body temperature and concomitant medications. AEs included solicited local and systemic events. Solicited local AEs were pain, tenderness, erythema/redness and induration/swelling, and solicited systemic AEs were fever, headache, malaise/fatigue, myalgia, vomiting and diarrhea. The unsolicited AEs were also checked using a diary card for 21 days after vaccination. The grade of AEs was assessed by the subjects using a standard scale according to FDA guideline: Toxicity Grading Scale for Healthy Adults and Adolescent Volunteers in Preventive Vaccine Clinical Trial Citation19 and MFDS guideline: Severity assessment of adverse event in vaccine clinical trials. If the subjects experienced SAEs, they were asked to report it for up to 6 months after vaccination. AE was sub-classified as an ADR according to the causality. ADR was defined as an AE suspicious of a causal relationship between the vaccine and the reaction. The causality was assessed by the investigators of each hospital.
Statistical analysis
Immunogenicity analyses were performed on the per-protocol set (PPS), which included all enrolled subjects who fulfilled visit 3 without any major deviation. Safety analyses were performed for all subjects exposed to study vaccines (full-analysis set, FAS). A total of 1,500 subjects (1,200 subjects 19–59 years of age and 300 subjects ≥60 years of age) were planned to be enrolled. The sample size was estimated based on the results of the prior clinical trial (phase I and II) with NBP607-QIV and phase III clinical trial with NBP607. Non-inferiority margin was set to 1.5 and 10% for adjusted GMT and seroconversion rate respectively according to the Guidance for Industry: Clinical Data Needed to Support the Licensure of Seasonal Inactivated Influenza Vaccine.Citation20 Non-inferior immunogenicity of NBP607-QIV vs. NBP607-TIV for shared strains was demonstrated if the upper limit of the 2-sided 95% confidence interval (CI) for the GMT ratio of NBP607-TIV/NBP607-QIV did not exceed 1.5, and the upper limit of the SCR difference (NBP607-TIV minus NBP607-QIV) did not exceed 10%. The sample size was estimated to provide at least 80% overall power to examine non-inferiority for all four strain comparison.
Statistical analysis was performed using SAS software (version 9.3 or higher; SAS Institute). Immunogenicity data were expressed in terms of adjusted GMTs (mean ± standard deviation) and CHMP criteria with two-sided 95% confidence intervals. The adjusted GMTs were estimated using an Analysis of Covariance (ANCOVA) model with baseline HI titer as covariate. Two-sided 95% CIs for GMTs and GMRs were calculated using the normal approximation of log-transformed titers, and percentages were calculated with approximate or exact 95% CIs. Safety data were described as the proportion of study subjects reporting local and systemic adverse reactions and severity expressed by the grade. The Chi-square test or the Fisher's exact test was used to analyze categorical variables. Results were considered statistically significant if the p value was less than 0.05.
Ethical approval
The study was performed with approval of the Institutional Review Board from each of the ten university hospitals. Written informed consent was obtained from all participants.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors appreciate the NBP607-QIV study team of SK Chemical Co., Ltd. and all the participants. The authors also thank the laboratory staffs and clinical research nurses of each participating hospitals.
Funding
This study was supported by SK Chemicals Co., Ltd and the Korea Healthcare Technology R&D Project of the Ministry of Health & Welfare of the Republic of Korea (no. A103001). This study was designed, executed, and analyzed by the sponsor. Although the sponsor reviewed a draft, the opinions expressed by the sponsor may not necessarily be reflected.
References
- Reed C, Meltzer MI, Finelli L, Fiore A. Public health impact of including two lineages of influenza B in a quadrivalent seasonal influenza vaccine. Vaccine 2012; 30:1993-8; PMID:22226861; https://doi.org/10.1016/j.vaccine.2011.12.098
- Belshe RB, Coelingh K, Ambrose CS, Woo JC, Wu X. Efficacy of live attenuated influenza vaccine in children against influenza B viruses by lineage and antigenic similarity. Vaccine 2010; 28:2149-56; PMID:20003926; https://doi.org/10.1016/j.vaccine.2009.11.068
- Ambrose CS, Levin MJ. The rationale for quadrivalent influenza vaccines. Hum Vaccin Immunother 2012; 8:81-8; https://doi.org/10.4161/hv.8.1.17623
- Belshe RB. The need for quadrivalent vaccine against seasonal influenza. Vaccine 2010; 28 Suppl 4:D45-53; PMID:20713260; https://doi.org/10.1016/j.vaccine.2010.08.028
- World Health Organization. Recommended composition of influenza virus vaccines for use in the 2012–2013 northern hemisphere influenza season. Weekly Epidemiological Record 2012; 87(10):83-95
- Doroshenko A, Halperin SA. Trivalent MDCK cell culture-derived influenza vaccine Optaflu (Novartis Vaccines). Expert Rev Vaccines 2009; 8:679-88; PMID:19485748; https://doi.org/10.1586/erv.09.31
- Abelin A, Colegate T, Gardner S, Hehme N, Palache A. Lessons from pandemic influenza A(H1N1): the research-based vaccine industry's perspective. Vaccine 2011; 29:1135-8; PMID:21115061; https://doi.org/10.1016/j.vaccine.2010.11.042
- Choi WS, Kim WJ, Cheong HJ. [The evaluation of policies on 2009 influenza pandemic in Korea]. J Prev Med Public Health 2010; 43:105-8; PMID:20383042; https://doi.org/10.3961/jpmph.2010.43.2.105
- Emborg HD, Krause TG, Hviid A, Simonsen J, Molbak K. Effectiveness of vaccine against pandemic influenza A/H1N1 among people with underlying chronic diseases: cohort study, Denmark, 2009–10. BMJ 2012; 344:d7901; https://doi.org/10.1136/bmj.d7901
- Song JY, Cheong HJ, Lee J, Woo HJ, Wie SH, Lee JS, Kim SW, Noh JY, Choi WS, Kim H, et al. Immunogenicity and safety of a cell culture-derived inactivated trivalent influenza vaccine (NBP607): A randomized, double-blind, multi-center, phase 3 clinical trial. Vaccine 2015; 33:5437-44; PMID:26314625; https://doi.org/10.1016/j.vaccine.2015.08.030
- Graaf H, Faust SN. Fluarix quadrivalent vaccine for influenza. Expert Rev Vaccines 2015; 14:1055-63; PMID:26098443; https://doi.org/10.1586/14760584.2015.1057573
- Suryadevara M, Domachowske JB. Quadrivalent influenza vaccine in the United States. Hum Vaccin Immunother 2014; 10:596-99; https://doi.org/10.4161/hv.27115
- Block SL, Yi T, Sheldon E, Dubovsky F, Falloon J. A randomized, double-blind noninferiority study of quadrivalent live attenuated influenza vaccine in adults. Vaccine 2011; 29:9391-7; PMID:21983154; https://doi.org/10.1016/j.vaccine.2011.09.109
- Greenberg DP, Robertson CA, Noss MJ, Blatter MM, Biedenbender R, Decker MD. Safety and immunogenicity of a quadrivalent inactivated influenza vaccine compared to licensed trivalent inactivated influenza vaccines in adults. Vaccine 2013; 31:770-6; PMID:23228813; https://doi.org/10.1016/j.vaccine.2012.11.074
- Bart S, Cannon K, Herrington D, Mills R, Forleo-Neto E, Lindert K, Abdul Mateen A. Immunogenicity and safety of a cell culture-based quadrivalent influenza vaccine in adults: A Phase III, double-blind, multicenter, randomized, non-inferiority study. Hum Vaccin Immunother 2016; 12:2278-88; https://doi.org/10.1080/21645515.2016.1182270
- Kieninger D, Sheldon E, Lin WY, Yu CJ, Bayas JM, Gabor JJ, Esen M, Fernandez Roure JL, Narejos Perez S, Alvarez Sanchez C, et al. Immunogenicity, reactogenicity and safety of an inactivated quadrivalent influenza vaccine candidate versus inactivated trivalent influenza vaccine: a phase III, randomized trial in adults aged >/=18 years. BMC Infect Dis 2013; 13:343; PMID:23883186; https://doi.org/10.1186/1471-2334-13-343
- Levandowski RA, Gross PA, Weksler M, Staton E, Williams MS, Bonelli J. Cross-reactive antibodies induced by a monovalent influenza B virus vaccine. J Clin Microbiol 1991; 29:1530-2; PMID:1885750
- Note for Guidance on Harmonisation of Requirements for Influenza Vaccines (CPMP/BWP/214/96). London, UK: Committee for Proprietary Medicinal Products (CPMP), 1997
- Guidance for Industry: Clinical Data Needed to Support the Licensure of Pandemic Influenza Vaccines. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research, 2007
- Guidance for industry: clinicaldata needed to support the licensure of trivalent inactivated influenza vaccines. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research, 2006