
ABSTRACT
Over the last decade the field of cancer biology has gained considerable data on genomic heterogeneity. This situation creates challenges and possibly opportunities for cancer treatment. The evolution of the tumor at all stages also requires the growing malignancy to confront and avoid the immune system. What we describe here is the interaction of two immune phenomena that work together to change the characteristics of the tumor, i.e., antigenic competition and immune editing. These two systems are mutually functional and their interaction is capable of altering the characteristics of the tumor for protection and survival in an immune competent host as well as restricting the diversity of the tumor clones. Therefore, the final outcome of these interactions can also become the key to the backdoor into the castle. Through an additional immune manipulation, autologous tumor cell immunization, we can achieve prevention of disease recurrence after surgical resection and by analyzing induced human monoclonal antibodies to the neoantigens, gain in site into the restriction of diversity of the mutant clones. These findings may also open the door for a pathway to immune prevention of cancer.
Introduction
Neoplastic or dysplastic cells are common. Based on autopsy studies, a “perfect” diagnostic test for breast cancer would detect disease in at least 10% of women who die from other causes.Citation1 Additionally, prostate cancer cells are found in 40% of men over the age of 60 and 60% over 80.Citation2 Yet the rates of invasive breast and prostate cancer requiring treatment are much lower than these autopsy studies would suggest. How do we explain the commonality of neoplasia, and the relative scarcity of invasive disease, based on these experiments of nature? The answer may be a consequence of multifaceted, but major components, of a highly evolved immune system. It is these collective efforts of the immune system and some of the ramifications that we intend to highlight in this publication.
Considerable attention is being given to immunotherapy as an essential means of augmenting our innate and adaptive immune capabilities to combat neoplastic disease and reduce the cost of treating advanced cancer. With respect to active specific immunotherapy (ASI), over 2 decades of clinical research, using a variety of compositions of cancer vaccines to treat advanced disease, have only led to incremental improvements.Citation3 A recent change in strategy, targeted reversal of tumor immunosuppression (e.g., checkpoint inhibitors) has also achieved a degree of clinical success in advanced disease patients.Citation4
Spurred by this recent clinical success, the Obama administrations got involved in cancer treatment and has allocated additional funds for a “Moon Shot” approach with significant attention paid to precision therapy. Yet if we do not understand the limits and restrictions inherent to the biology of cancer we risk wasting valuable resources. These approaches are severely hamstrung by the genomic heterogeneity of malignant disease.Citation5
Recently, Ling and colleaguesCitation6 evaluated a single, approximately 3.5 cm squared hepatocarcinoma by sequencing or genotyping nearly 300 regions from the tumor. They estimated nearly 100 million coding region mutations would be found across the entire sample. It leads one to believe that with a few biopsies, neoantigen discovery intended to represent the totality of a patient's tumor will be extremely difficult, if not impossible. They estimated drug resistance to be 1 in 5000 tumor cells of any individual clone. This high probability of drug resistance creates paradoxes that make targeted therapies in solid tumors problematic. It is now an established fact that adenocarcinomas are multi-clonal with inter- and intra-genomic heterogeneity. The dynamic range of heterogeneity between tumors is still unclear, however the rapid advances in the molecular characterization of tumors, including gene sequencing has driven the precision medicine approach to treatment. Still, this approach of identifying a mutational product from the tumor genome and using it to target drugs, immune cells or antibodies is a potential, but less effective paradigm of research and/or drug development. Despite the enthusiasm surrounding rare cases of success, most patients with advanced cancer do not benefit from the precision strategy, nor has this approach, to date, been shown to improve outcomes in controlled clinical trials.Citation7
Outside-in vs. inside-out strategies
If we intend to leverage the power of the immune system for cancer treatment, we must adopt a viewpoint that includes the host-tumor interactions. The approach now involves identifying key genetic lesions from one or a small number of biopsies evident on the genomic level and extrapolating outward assuming many of these unique markers are translated from the genetic sequence to the protein level to a meaningful degree and distributed or present in other tumors of the same histological type with a degree of homogeneity. We can refer to this as the “inside out” approach. Also, this translation must happen in a manner compatible with active immune recognition. Wood et al.,Citation8 in 2007, demonstrated that this is likely a false premise, among pairs of colon tumors, within the array of mutant clones less than 2 to 3 mutations were shared between patients. Furthermore, no guarantees were provided regarding the intratumoral incidence or distribution of these shared genetic lesions within a given tumor. Thus, identifying a common target by genomic sequencing of the tumor cells or from biopsies is a dangerous assumption not yet borne out by biological reality inherent to the disease.
Our 30 years of perspective of patient specific active immunotherapy convinces us that amplifying the patient's immune response, arming it with the appropriate tools and relevant targets, will simultaneously increase the patient's inherent immunity to the majority of the foreign neoantigens of the tumor cells. This provides for the patient's immune system to identify the relevant tumor antigens, and profile and decode how a patient's own immune system views the biological challenge of malignancy. If we truly understand the mechanisms by which these tumors arise, simultaneously co-opting and subverting the healthy immune system, we can work toward eliminating these blind spots for clinical gain. This will benefit the clinical outcome by destroying tumors and stave off immunosuppression. This will also benefit most of cancer patient's. We will refer to this as the “outside in” approach, and can achieve it by immunizing the patient with the autologous tumor for the purpose of establishing systemic immunity and preventing recurrence of disease.
Host-tumor interaction: Immune editing of tumors
Experimentation examining the nature of adenocarcinoma development has uncovered an inverse correlation between immune competence and adenocarcinoma development. But, the relationship between immune function and malignant disease is complex and not all responses are curative. For example, according to the “immune editing” hypothesis,Citation9,10 poorly immunogenic tumors, which escape immune surveillance, may actually be created by normal immune function through a long-term process of clonal selection. Immunogenic clones which arise over time, are appropriately recognized by the immune system and pruned away, thus the tumor that escapes control and becomes detectable has specifically evolved to avoid competent immune recognition. The complex relationship between immune function and cancer development is undeniable. It is clear that the clones of tumor that survive immune editing are poorly immunogenic and can continue to propagate, even in an immunocompetent host.
In these studies, transplanted tumors in immunocompetent mice were shown to be qualitatively different from tumors in immunodeficient mice. This observation, which led to the formulation of the cancer immune editing hypothesis, is based on comparative analyses of carcinogen-induced tumors harvested from immunocompetent and immuno- deficient mice. In these experiments, tumor cell lines were established from tumors arising in each group of mice, and these cells were then injected into immunodeficient recipient mice or immunocompetent wild-type (WT) recipient mice. Tumor cells from carcinogen-treated WT mice formed progressively growing tumors in both immunodeficient mice and naïve syngeneic immunocompetent mice 100% of the time. In contrast, although tumor cells from carcinogen-treated immunodeficient mice grew progressively when transplanted into immunodeficient mice only half of the tumor cell lines were capable of forming progressively growing tumors in naïve syngeneic immunocompetent recipients whereas the other half of the cell lines were rejected by the recipients. Thus, tumors from immunodeficient mice are termed “unedited” and further designated as “progressor” or “regressor” to denote their growth phenotypes after injection into naïve WT recipients. Carcinogen-induced tumors from immunocompetent mice are termed “edited” because they are less immunogenic and show only a progressor growth phenotype.
While the immune editing theory is a powerful model for cancer development, how poorly immunogenic clones remain during the equilibrium phase has yet to be defined. Is it simply a function of immune tolerance or possibly a function of the PD1/PDL1 axis? We intend to expand on this model with another immune mechanism: antigenic competition.
Host-tumor interaction: Antigenic competition
The reduced immune response to an antigen as a result of a previous, closely spaced reaction to a different, stronger antigen has been described.Citation11,12 This form of immune suppression, is generally referred to as “antigenic competition.”
Our studies suggested that a competition that had occurred during priming mainly affected the development of the immunocompetent, progenitor cell compartment.Citation13 The primary and secondary hemagglutinin response levels in the sheep red blood cell (SRBC)/rat red blood cell (RRBC), combination challenges are shown in . The 2-mercaptal ethanol (ME) resistant antibody was assumed to represent 7S hemagglutinin (the specific IgG) and the difference (total antibody minus 2-ME resistant antibody was considered to be 19S (the “natural antibody” IGM). In this competition format a suppressed primary hemagglutinin profile was measured along with a feeble or absent, antigen specific IgG response (). These animals all had a reduced secondary response that was mainly IgG antibody. In all 3 groups the suppressed secondary response was not comparable in detectable serum antibody to a normal primary response achieved with a similar dose of antigen. However, the antibody profile was characteristic of normal secondary response profiles in that there was an early rise in titer (free antibody detected at 2-days). Thus, an intravenous dose of 1 ml of 1% SRBC administered 2 d previously is capable of markedly suppressing a RRBC primary and secondary hemagglutinin response, even when a 10-fold greater dose of RRBC is injected.Citation13
Figure 1. Comparison of total (O) and 2 MEresistant (A) anti-rat hemagglutinin levels during the primary and secondary immune response in mice injected with RRBC alone, and total (o) and 2 ME-resistant (A) anti-rat hemagglutinin levels during the primary and secondary immune response in experimental mice immunized with RRBC 2 d after an injection of 1 ml of 1.0% SRBC: a, 1 ml of 0.1% RRBC on day O and day 20; b, 1 ml of 1.0% RRBC on day O and day 20; c, 1 ml of 10.0% RRBC on day O and day 20. Each point represents the mean titer.
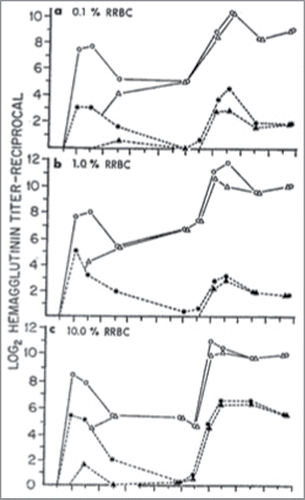
The exact primary and secondary hemagglutinin profile results were measured when we used a SRBC and aggregated Human gamma globulin format of antigen competition. In the antigen competition systems that we studied there is a reduced primary immune response to the second antigen. Corresponding to this suppressed primary response is a deficient secondary immunologic capacity. In all cases the secondary response is below a normal primary hemagglutinin response, in spite of the fact that the IgG/IgM profiles are characteristic of a secondary hemagglutinin response. Also, the fact that a primary response to a different antigen (HGG) can be obtained in animals, which at the same time are incapable of producing a normal secondary response to RRBC, indicates that the latter is mainly a lack of sensitized immune progenitor cells. It was also demonstrated that increasing the dose of the secondary challenge could reduce the degree of suppression of the anamnestic response. These results suggested that under certain conditions the secondary immune response could consist of both reacting sensitized and uncommitted immunocompetent cells.
Antigen competition can be summarized as follow: it occurs when exposure to a strong antigen/immunogen, precedes a weaker immunogen by 1 to 3 days; for the weaker antigen there is little to no primary humoral immune response and the secondary immune response may require participation of both sensitized (memory) cells and uncommitted immunocompetent cells. Antigen competition is not associated with the antigen and dendritic reticular cell localization function; the deficiency in a competition system is attributable to an impairment that ultimately affects the immunocompetent (progenitor) cell or unit.
Recent studies of antigenic competition have demonstrated these same principals with respect to cell mediated immunity following acute challenge with a viral vaccine.Citation14 In terms of scale, while intradermal vaccinations with the colon cancer or viral vaccines elicit immediate systemic effects, it is likely that long-term chronic challenge over the timescale of many years can have devastating local effects (tumor clone selection) which lead to future immune tolerance aiding disseminated disease. It is reasonable to speculate that antigenic competition is a stable part of the immune surveillance to have fully functional availability and distribution of resources from a limited compartment of progenitor immunocompetant cells to deal with a plethora of bombarding exogenous antigens.
Immune editing-driven by antigenic competition
We can now hypothesize on the networking of these two host-tumor interactions and understand how they interact to produce a tumor that can survive and grow in an immune competent host. Also, if our speculation is correct, it clarifies the success of the autologous tumor, outside-in approach to cancer vaccine active specific immunotherapy. A schematic of the two host tumor interactions in tumor cell populations is shown in ().
Figure 2. A speculative schematic of antigen competition driven immune editing during the evolution of an adenocarcinoma. At the outset we assume a normal curve of immunogenic clones ranging from weak to strong immunogenicity. Antigen competition driven effects would, through immune editing, begin to create an expanding population of weakly immunogenic, or immune tolerant clones while minimizing the moderate to strongly immunogenic clones. In time these immune tolerant clones can survive and prove fatal in an immune competent host.
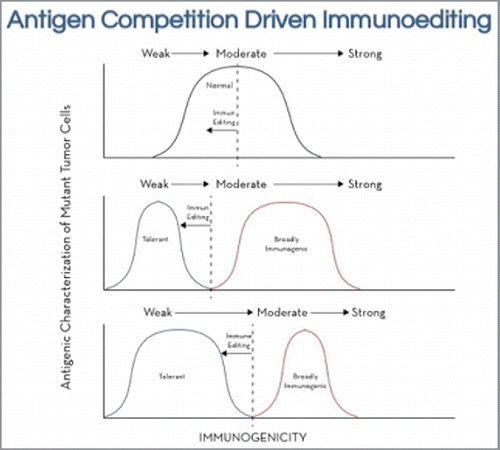
It is important to emphasize that the interaction is a dynamic process and proceeds while the tumor is developing or evolving over years. The burgeoning tumor with a myriad of immunogenic phenotypes, capable of mutating randomly, initially displays antigens with a normal distribution of immunogenicity. This is especially likely if the mechanism for mutational generation is driven by non-Darwinians means, which has been suggested by compelling bioinformatics analysis.Citation6 Due to antigenic competition, the immune system prioritizes the strongest antigens. The moderate to strong immunogenic phenotypes being continuously eliminated, and through immune editing, the tumor is sculpted with a preponderance of tumor cells to which the immune system appears to be tolerant. These tumor cells are capable of surviving and eventually becoming fatal to an immunocompetant host. The results are three-fold: 1) the remaining clones are weakly immunogenic, 2) the host immune system has become tolerant to these tumor cells and 3) there is potentially restriction of the diversity of the clones. This last point, the restriction of diversity of the clones is of special interest. It will pay to concentrate on the less heterogeneous aspect of the tumor derived from antigenic competition driven immune editing.
While numerous observations have been made in humans and animal models, the most clinically relevant examples are evident in the field of vaccine development. If a multivalent vaccine is required to address a plurality of antigens (i.e., various isotypes of botulism or influenza), the antigenicity of the individual components must be balanced to generate an effective immune response against each antigen. On the one hand, antigenic competition can be viewed as an important immunologic failsafe. The immune system should be preoccupied with strong more likely foreign antigens to prevent viral and bacterial infection and limit potential autoimmunity. While this gambit has proven evolutionarily successful, it is not perfect and may represent a “backdoor” for cancer development to subvert a healthy immune response and invade the castle.
Scientists have known for some time that the immune system while powerful has limitations. With respect to cancer, these observations have important clinical ramifications. If strongly antigenic mutants precede the creation of weaker clones, little to no primary humoral or cellular immune responses may be generated toward the latter. Furthermore, any future secondary immune response against these remaining clones may require participation of other sensitized (memory) cells and uncommitted immunocompetent, progenitor cells. Only recently, with the discovery of immunosuppressive tumor microenvironments and the process of immunoediting, have we discussed the limitations, or inherent “back doors,” for the immunological creation of a developing malignancy. While the elimination phase of immune editing is difficult to prove directly, its existence is highly likely and formed the basis of the original immune surveillance theory. Furthermore studies of tumor development in the pancreas have estimated malignant disease can take 20 years to develop and other estimates for and other adenocarcinomas are 1% to 10% of a lifetime. This fits the equilibrium phase of the immune editing theory quite well.
This mechanism potentially addresses a current paradox within the immune editing theory. There is no doubt that the establishment of an immunosuppressive tumor microenvironment is a hallmark of advanced malignant disease. However, if the earliest immunosuppressive microenvironments are too strong, the process of immune editing could not occur. The cells charged with the sculpting of this poorly immunogenic population would themselves be suppressed. Therefore, when does the critical breakpoint between immunopermissive and immunosuppressive occur?
If immune editing is driven by antigenic competition, poorly immunogenic clones can be accumulated in a long enough period of time within and immunopermissive enviornment. Their selection is simply dictated by the strength of their antigenicity. Once the burgeoning tumor has been sufficiently immune edited from an antigenic standpoint and these cells have acquired mutations or signal transduction/dysfunction sufficient to establish an immunosuppressive environment (ie., Treg cells, expression of the PD1/PDL1 axis, etc), enhanced migration and unlimited replicative potential, the disease progresses to the escape phase, armed with the tools necessary to extravasate from its original location to colonize the rest of the body at will. Consequently, metastatic disease is the end result of a lengthy stepwise process of genetic and immunologic pressures with many factors at play.
The clinical implications of antigenic competition-driven immune editing
A major factor in designing effective clinical immunotherapy trials with the “outside in” approach is patient status with respect to disease stage, the dilemma of timing, composition and dose of the immune challenge. These factors were first studied using a transplantable, syngeneic L-10 hepatocarcinoma in Strain 2 guinea pigs.Citation15,16
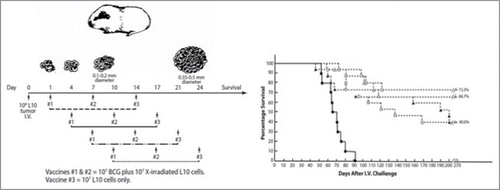
These animals were inoculated with live tumor cells on day 0, to create a stage of metastasis and were immunized 3 times with a tumor cell plus Mycobacterium bovis (TICE BCG) vaccine beginning on days 1, 4, 7 or 10. The subsequent efficacy of treatment was inversely proportional to the time of treatment and, by extension, degree of tumor burden (). The optimized vaccine induced active specific immunotherapy, and provided immune protection against submicroscopic metastasis (0.1–0.2mm) or minimal residual disease. Tumor nodules in the lung at the 0.35 to 0.5 mm diameter were not cured. Tumor nodules at both of these size ranges are barely visible under the microscope. This clearly suggests the optimum use of active immunotherapy is for treating occult disease. The difficulties of previous pre-clinical and clinical immunotherapeutic is approaches, with respect to timing, patient selection, and composition, should be viewed through the prism of these results. However, if antigenic competition is the driving mechanism for the equilibrium phase of immune editing, we could predict these results. There is a significant period of time where functional immunological access within the tumor microenvironment can be augmented for clinical gain.
Figure 3. Experimental studies of active specific immunotherapy in the guinea pig tumor model system: Percentage survival as a function of time after challenge with 106 L10 cells i.v. Vaccinations 1 + 2 = 107 BCG + 107 L10; 3: L10 alone. (•) control; (O) 3 vaccinations, days 1, 7, 14; (Δ) 3 vaccinations, days 4, 10, 17; (ם) 3 vaccinations, days 7, 14, 21; ( ▴ ) 3 vaccinations, days 10, 17, 24.
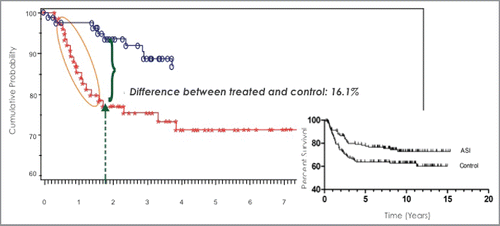
Based on these preclinical studies, active, patient specific immunotherapy of occult disease achieved major clinical success in stage II (T3 &T4a, b) colon cancer and is adaptable to many other forms of adenocarcinomas. Clinical studies with a patient specific vaccine called OncoVAX, has been performed in hundreds of colon cancer patients.Citation17,18 This programmed approach has been shown in clinical trials to possess the 4 P's necessary for patient specific active immunotherapy, Personalized, Precision, Potency and Prevention of post surgical occult disease (). The significant prevention of disease progression was seen over a 15-year follow-up of recurrence-free interval and recurrence–free survival.Citation19
Figure 4. Recurrence-free interval (RFI) in Stage Il colon cancer of OncoVAX treated versus surgery tumor resection alone control patients. The latter is standard of care for Stage Il colon cancer, and these patients are classified as an unmet medical need. The proportion of recurrence-free patients at 4 y was 74% for non-treated patients and 88% for OncoVAX treated patients. The significant difference between the 2 groups was p = 0.011 and the recurrence free survival was p = 0.032. There was a 16.1% difference in recurrences at 1.8 y median follow-up. It takes 3.0 g of tumor or greater to successfully prepare an autologous tumor vaccine with >90% cell viability and sterility, thus these patients were in the higher risk T 3 and T4 A&B pathological classification. To avoid bias, randomization was performed after each enrolled patient had a prepared vaccine that met specification. There was not just a decease in the number of recurrences in the OncoVAX treated patients but also difference in the rate of recurrence. A recent analysis of the long-term survival impact of these results on the remaining patient population at 15 y is shown (right inset).
The humoral response to autologous colon tumor immunization in OncoVAX treated patients may shed more light on this issue. In the Phase III trial we isolated B-cells from these patients and successfully produced a variety of human monoclonal antibodies (HuMab) specific to tumor associated antigens. We speculated that at some point during the course of immunization, which was designed primarily for stimulation of a strong cell-mediated immune response, there might be a transient humoral immune response of some magnitude. In animal models, humoral immunity is present during the induction of cell-mediated immunity.
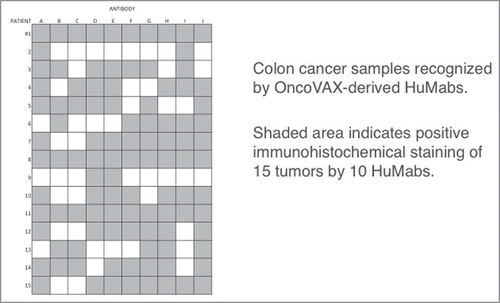
This was the correct strategy to achieve HuMab(s) with demonstrated specificity and stability. In our studiesCitation20 the most productive fusions were achieved with B-cells taken during a one-week window during the induction course of the immunizations. An extensive in vitro screening was performed on these monoclonal antibodies. The individual monoclonal antibodies usually reacted homogeneously by histochemistry, with the tumor as determined with either cryostat or paraffin sections. The pattern of reactivity of 10 of the human monoclonal antibodies to histological sections of colon adenocarcinomas from 15 patients is shown in . The matrix of reactivity of antibodies reacted with between 47% and 80% of the tumor specimens. No single antibody reacted with all 15 tumors. In tissue sections from individual patients, the range of reactivity varied from reactivity to all 10 antibodies to reactivity to as few as 2 antibodies. Overall however, 70% of the antibodies reacted with 73% of the tumor sections. Also, by strategically selecting 2 antibodies (in HuMabs I and J), the overall reactivity to individual patient colon cancer sections, reactivity of one or the other antibody, was 14 out of 15 tumor sections, or 93%. These are surprising results that indicate that in contrast to some of the genomic sequencing data, there may at some stage of tumor development, been a restriction of the diversity of the immunogenic tumor clones. We are now moving to fully characterize the neoantigens associated with these HuMabs.
Figure 5. Distribution of antigens in paraffin sections of colon tumors. Shaded area indicates positive indirect positive indirect immunoperoxidase staining of 15 tumors by 10 OncoVAX treated patient derived human monoclonal antibodies. None of the HuMabs tested were derived from any patients whose tumor samples were tested.
Thus, an improved method for identifying immunogenic tumor-specific antigens is based on an outside-in approach. A more meaningful understanding of immune interaction with human tumors, should start from the immune system's perspective to capture the full diversity of potential antibody and/or T-cell targets. Consequently, these studies uniquely exploit the human immune system to determine which targets within the proteomic milieu are simultaneously cancer-specific and immunologically relevant. By definition, because this vaccination process is autologous, antigens defined as “self” are inherently ignored and disease-specific mutations, protein-protein interactions, and post-translational modifications become the immediate immunological focus. This “outside-in” approach is a method for using the agnostic immune response to create a molecular database of functional cancer-specific immunogens.
Our thesis is that antigen competition driven immune editing creates a restricted set of poorly immunogenic clones, possibly because the tumor cells were too similar to normal cells from which they were derived and these cells can survive in an immunocompetent host because of immune tolerance. One interesting question then is, when we perform patient specific active immunotherapy by a vaccine consisting of these excised and live, metabolically active, tumor cells and BCG, administered intradermally, are we breaking tolerance? This is a reasonable assumption based on the intradermal injection of these tumor cells in an environment replete with educated antigen presenting dendritic reticular cells (DCs). These DCs, which are concentrated in the epidermis, previously called Langerhans cells, are central regulators of the balance between immunity and tolerance. Between the exposure of these weakly immunogenic tumor cell clones to a heterogeneous population of DC subsets, and the trafficking of immunocompetent lymphocytes to these vaccination sites along with the innate immunity stimulated, provides the conditions that can break tolerance. One additional component of antigen competition is the fact that in certain cases, while the adaptive immune response is blocked, there is still the establishment of memory cells to the second, weaker antigens. When we break tolerance, we assume that both the memory cell compartment and the uncommitted immune competent cells are involved. This may relate to the long (15 year) duration of the recurrence-free survival and –interval, clinical benefit in the described phase III colon cancer trial.
Conclusion
With current advances in sequencing and immune-based host-tumor interactions, we have an opportunity to gain a better understanding of how cancer evolves. If antigenic competition is the engine for driving immune editing, this provides clinical opportunities and boundaries. If we seek to advance the treatment and prevention of cancer, we must accept the limitations established by its natural evolution. Successful systemic education and destruction of micrometastases, truly minimal residual disease, provides an excellent example for additional opportunities.
As outlined above, an improved approach for identifying immunogenic tumor-specific antigens is based on adopting an “outside-in” approach. Any meaningful understanding of immune interaction with human tumors, should start from the immune system's perspective to capture the full diversity of potential antibody and/or T-cell targets. This is the preferred means of discovering targets within the proteomic milieu, which are simultaneously cancer-specific and immunologically relevant. By definition, because this vaccination process is autologous, antigens defined as “self” are inherently ignored and disease-specific mutations, protein-protein interactions, and post-translational modifications should become the immediate immunological focus.
While the most recent success in treating advanced disease with immunological approaches is exciting, it is logical that we should not have to wait for recurrence or progression of disease to treat cancer. There is much to be gained, with respect to current lives and developing future treatments, by staying committed to achieving sustained recurrence-free survival for most cancer patients following surgical resection. We find ourselves in an exciting time in cancer immunotherapeutic research. Through the diligent work of many, we are finally establishing a complete and comprehensive picture of how cancer emerges and evolves with respect to immune function. This renaissance is occurring in parallel with advancements in technology designed to identify and exploit these pathways for clinical gain. As a field, we must grasp this opportunity and shift our focus away from just curing cancer, but relegating malignant disease to the same designation as smallpox and polio: problems that simply went away.
Disclosure of potential conflicts of interest
MGH, Jr. is Co-Founder and Chairman Emeritus of Vaccinogen, Inc.
Acknowledgment
The authors would like to acknowledge Brian Manning for his analogy of describing antigenic competition as a system access point which cancer can exploit.
References
- Welch HG, Black WC. Using autopsy series to estimate the disease “reservoir” for ductal carcinoma in situ of the breast: how much more breast Cancer can we find? Ann Internl Med 1997; 127 (11):1023-28; https://doi.org/10.7326/0003-4819-127-11-199712010-00014
- Zlotta AR, Egawa S, Pushkar D, Govorov A, Kimura T, Kido M, Takahashi H, Kuk C, Kovylina M, Aldaoud N, et al. Prevalence of Prostate cancer on autopsy: cross-sectional study on unscreened Caucasian and Asian men. 2013. J Natl Cancer Inst 2013; 17 105 (14):1050-58; https://doi.org/10.1093/jnci/djt151
- Kudrin and Hanna. Overview of the cancer vaccine field: Are we moving forward? Hum Vaccin Immunother 2012; 8:1135-44; PMID:22854670; https://doi.org/10.4161/hv.20474
- Pardoll DM. The blockade of tumor checkpoints in cancer. Nat Rev Cancer 2012; 12:252-64; PMID:22437870; https://doi.org/10.1038/nrc3239
- Dimond PF. Intratumor heterogeneity poses challenge for cancer “moonshot” program. Gen Exclusive 2016; 36(5).
- Ling S, Hu Z, Yang Z, Yang F, Li Y, Lin P, Chen 1<, Cao L, Tao Y, Hao L, et al. Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. PNAS 2015; 112:E6496-6505; PMID:26561581; https://doi.org/10.1073/pnas.1519556112
- Tannock IF, and Hickman JH. Limits to Personalized Cancer Medicine. N Engl J Med 375 2016; 13:1281-94.
- Wood L. The Genomic Landscapes of Human Breast and Colorectal Cancers. Science 2007; 318:1108-13; PMID:17932254; https://doi.org/10.1126/science.1145720
- Dunn GP, Old 1J, Schreiber R. The Immunobiology of Cancer Immunosurvellance and Immunoediting Immunity. Aug 2004; 21 (2):137-148.
- Schreiber RD, Old LJ, Smyth MJ. Cancer Immunoediting: Integrating immunity's roles in cancer suppression and promotion. Science 2011; 331:1565-70; PMID:21436444; https://doi.org/10.1126/science.1203486
- Adler FL. Antibody formation after injection of heterologous immune globulin Il Competition of Antigens. J Immun 1957; 78 (3):201-10; PMID:13429087
- Eidinger D, Khan SA, Millar KG. The effect of antigenic competition on various manifestations of humoral antibody formation and cellular immunity. J Exp Med 1968; 128:1183-200; PMID:5696281; https://doi.org/10.1084/jem.128.5.1183
- Hanna Jr„ MG, Peters LC. The effect of antigen competition on both the primary and secondary immune capacity in mice. J Immun 1970; 104 (1):166-177; PMID:4189099
- Muschaweckh A, Bucholz VR, Fellenzer A, Hessel C, Konig PA, Tao S, Roony T, Heikenwilder M, Bush DH, Korn T, et al. Antigen-dependent competition shapes the local repertoire of tissue-resident memory CD8+ T cells. JEM 2016; 213 (No. 13):3075-86; https://doi.org/10.1084/jem.20160888
- Hanna Jr. MG, Peters LC. Morphological and functional aspects of active-specific immunotherapy of estaqblished pulmonary metastasis in guinea pigs. Cancer Res 1981; 41:4001-6; PMID:7285011
- Fidler IJ, Hanna Jr. MG. New approaches to specific and nonspecific immunotherapy of established cancer metastasis. Published in Fundamentasl Mechanisms in Human Cancer immunotherapy, Saunders, Daniels, Serrou, Rosenfeld and Denny Eds, Elsevier North Holland, Inc., 1981, pps 425-437.
- VermorkenJB C, vanTinterenH G, Ezinga R, Meijer S, Scheper RJ, Meijer CJLM, Bloemena E, Ransom JH, Hanna Jr. MG, Pinedo H. Active specific immunotherapy for stage Il and stage Ill human colon cancer: a randomised trial. Lancet 1999; 353:345-50; PMID:9950438; http://dx. doi.org/10.1016/S0140-6736(98)07186-4
- Hanna Jr. MG, Howard J, Vermorken J. Active specific immunotherapy:Using tumor heterogeneity to successfully fight cancer. Hum Vaccines and Immunotherapy 2014; 10 (11):3286-96; https://doi.org/10.4161/hv.28886
- de Weger VA, Turksma AW, Voorham QJ, Euler Z, Bril H, van den Eertwegh AJ, Bloemena E. Clinical effects of adjuvant active specific immunotherapy differ between patients with microsatellite stable and micro- satellite instable colon cancer. Clin Cancer 2012; 18(3):882-889
- Haspel MV, McCabe RP, Pomato N, Janesch NJ, Knowlton JV, Peters LC, Hoover HC Jr., Hanna MG Jr.. Generation of tumor cell-reactive human monoclonal antibodies using peripheral blood lymphocytes from actively immunized colorectal carcinoma patients. Cancer Res 1985; 45:3951-61; PMID:4016762