
ABSTRACT
The evaluation of the immunogenicity of Sabin strain based Inactivated Poliovirus Vaccines (sIPV) necessitates the use of wild strains in neutralization assays to assess the potential cross-reactivity of antibodies. The live virus strains including wild and Sabin strains must be handled in level 3 biocontainment laboratories. To develop an alternative assay without the use of a live virus, we constructed Mahoney, MEF-1, and Saukett pseudovirions by inserting luciferase reporter genes into intact capsid proteins. Afterward, we developed a pseudovirus-based neutralization test (pNT) and evaluated for the specificity and reproducibility. We tested serum samples from a clinical trial on sIPV vaccines by pNT and compared the results with those obtained from conventional neutralization tests (cNT). A strong correlation was observed between two methods, with the correlation coefficients of all three types of IPV vaccines being greater than 0.82 (p < 0.0001). The Geometric Mean Titer (GMT) values obtained by pNT were approximately four times higher than that by cNT, revealing the better sensitivity of pNT. In conclusion, pNT is a safe, rapid and sensitive quantitative assay with the potential of being an alternative for the evaluation of the potency of polio vaccines.
Introduction
Poliomyelitis, caused by poliovirus, is an acute infectious disease that may lead to the acute flaccid paralysis in children. An enterovirus of the family Picornaviridae, poliovirus has three serotypes. The positive-sense, single-stranded RNA genome is comprised of a single open reading frame flanked by a 5ʹUTR region and a 3ʹpoly(A) tail. After its entry into the host cell, the virus employs cellular mechanisms to translate the polyprotein. The polyprotein is initially cleaved by the viral proteases such as 2Apro and 3Cpro into P1, P2, and P3 proteins, with P1 protein being further cleaved into VP1-VP4 capsid proteins, whereas P2 and P3 into functional proteins for viral replication and packaging.Citation1,Citation2
Live attenuated oral poliovirus vaccine (OPV, made with attenuated strains) has served as the primary tool to eradicate polio worldwide. However, the vaccine has the potential to result in rare adverse cases of vaccine-associated paralytic poliomyelitis (VAPP) and circulating vaccine-derived polioviruses (cVDPVs), which have become the major concern. According to WHO report, the global annual burden of VAPP is estimated at about 500 cases and total 96 cases of cVDPV occurred in the world last year.Citation3,Citation4 Even in high-income countries with all inactivated poliomyelitis vaccine (IPV, made with wild strains) immunization schedule, VAPP risks remain; Foiadelli et al. reported the first VAPP case in an immunodeficient infant detected in Albania, which was later confirmed to be a Sabin-like strain in Italy.Citation5 Clearly, phasing out OPV vaccination eventually is currently on the agenda by the international community. The World Health Organization (WHO) has set the goal of global polio eradication by 2018. For those countries still using the live vaccines, the previous trivalent oral polio vaccine (tOPV) was replaced by the bivalent vaccine (bOPV) in the routine immunization program, along with the introduction of at least one dose of the inactivated polio vaccine (IPV).Citation6-Citation10 Given that wild type virus must be handled in level 3 biocontainment laboratories, WHO encourages the development of novel polio vaccines such as IPV based on the attenuated Sabin strains (sIPV).Citation11 The first sIPV combined with DTaP was approved in Japan in 2012. China approved the stand-alone sIPV the following year, with ongoing development in more manufacturers.Citation12
The traditional IPV is derived from the wild virus strains (wIPV), mostly Mahoney, MEF-1, and Saukett for type 1, 2 and 3. During clinical evaluation of sIPV vaccine, it is important to determine the cross-protection of the vaccine against the wild virus, which is achieved by carrying out a neutralizing assay using wild-type virus strains (wt virus). However, the wt virus-based neutralizing assay poses a challenge to most OPV vaccine producers, since they are routinely operating the attenuated Sabin strains. In addition to vaccine evaluation in clinical trials, the neutralization test needs to be used in the quality control of IPV, i.e. potency test of final vaccine bulks in rats. Clearly, the use of the wt virus not only presents safety issues to the operators but also potentially results in environmental contamination. Indeed, WHO recommends that tight biosafety measures be taken to prevent leakage of live poliovirus.Citation13 Therefore, it would be desirable to develop new assays in which no live wt virus would be used.Citation9
Pseudoviruses are virus-like particles which can replicate for one cycle and are, therefore, considered to have no or minimal biosafety concerns. In recent years, pseudoviruses have been used in place of some wt viruses in neutralizing assays which otherwise must be conducted in level-3 biocontainment laboratories. These viruses include SARS-CoV, H7N9, and MERS.Citation14-Citation16 In this study, three pseudoviruses for wild Mahoney, MEF-1, and Saukett strains were constructed and used to develop a neutralization test in place of wt viruses. Here, we report that the new assay is superior to the traditional assays in terms of reproducibility and sensitivity; it could be a viable alternative neutralizing assay for potency analyses of polio vaccines.
Results
Construction and characterization of polio pseudoviruses
The polio pseudoviruses investigated in this study include Mahoney, MEF-1, and Saukett types, all of which were generated by transfection with vectors of their respective capsid proteins and replicon RNA.
As shown in , 293FT cells transfected with capsid protein vectors demonstrated apparent green fluorescence, indicating successful expression of the proteins. Harvested pseudoviruses were concentrated 50 times through ultrafiltration, and VP1 expression analyzed using western blotting (). The clear bands corresponding to VP1 (approximately 33kDa) in pseudovirus samples across different serotypes (lane 2) were observed as well as in the positive control, which as the sIPV monovalent bulks (lane 1). However, the molecular weight of the pseudo-Mahoney VP1 protein is visibly larger than that of the positive control as shown in Western blot. At this time we were unable to explain the observation. However, given the gene sequences have been verified and antigenicity confirmed, the difference in molecular weight is likely due to the cell substrates used to propagate the virus. Transmission electron microscopy analysis revealed that the morphological characteristics of pseudovirus particles across all serotypes resembled those of the true virus (20-30nm), suggesting that pseudoviruses were successfully packaged in the 293FT cells ().
Figure 1. Schematic view of PV capsid expression vector, replicon RNA, and the production of pseudovirus in 293FT cells. 293FT cells were first transfected with the pcDNA6.0-P1-EGFP plasmid to express P1 proteins, followed by transfection with the replicon RNA. As an encapsulated gene fragment, replicon RNA serves as a template for the synthesis of P2 and P3 proteins, whereas the 2Apro protease releases EGFP and luciferase and cleaves P1 into VP1-VP4 capsid proteins to facilitate pseudoviral packaging.
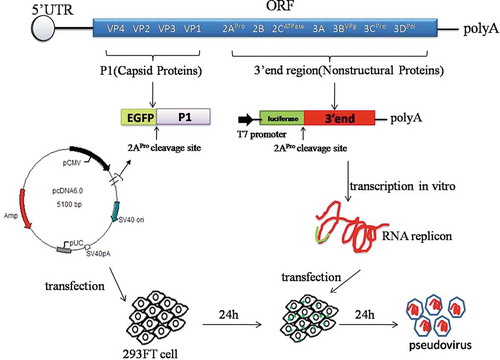
Figure 2. Characterization of pseudo-PVs. (a) Expression of pseudovirus capsid protein P1 and fluorescent protein EGFP fusion. 293FT cells transfected with capsid protein vectors produced green fluorescence, indicating successful expression of the proteins. (b) Western blot analysis for the verification of the expression of pseudovirus capsid protein VP1. Lane 1 is a positive control with monovalent sIPV vaccine corresponding to each pseudovirus type; lane 2 represents concentrated samples collected from Mahoney, MEF-1, and Saukett pseudovirus types; lane 3 is a negative control with supernatant harvested from 293FT cells. The antibodies used in the assay were monovalent rabbit antibodies against the virus and HRP-conjugated mouse anti-rabbit IgG. All three pseudoviruses and positive control show VP1 band. (c) Visualization of Mahoney, MEF-1, and Saukett pseudovirions using transmission electron microscopy: diameters are approximately 30nm, with the morphology similar to the wt virus (pointed by in-figure arrowheads). (d) Neutralization curves of PV pseudovirus with mouse anti-PV sera of three different types showed the type-specificity for the neutralization of pseudovirus (Each type serum was repeated three times).
In addition, we conducted separate neutralization tests using the mouse sera immunized with monovalent vaccines. As shown in , pseudoviruses were neutralized by the corresponding serum type. For the titration of pseudoviruses, a relative light unit (RLU) value two times greater than the value of the cell control was considered as positive. Based on these criteria, virus titers of Mahoney, MEF-1, and Saukett pseudoviruses prepared in this study were determined to be 1.3 × 107, 2.6 × 106, and 1.7 × 106CCID50/mL, respectively.
Development of pseudovirus neutralization assay
The determination of serum titers during the pseudovirus neutralization assay was based on the end-point determination;Citation17 specifically, it is the inverse of the highest serum dilution that inhibits 50% of infection.
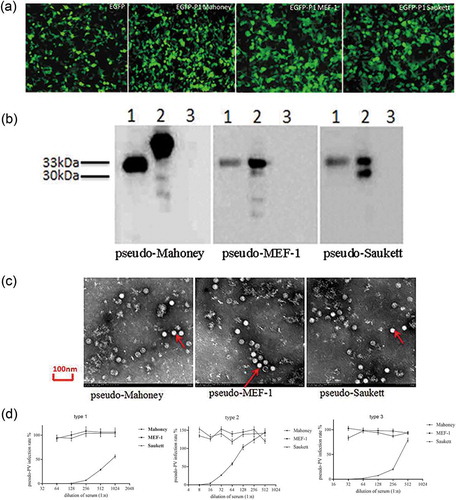
We first determined the linearity range of pseudovirus infection. As shown in , a linear response was observed with the use of the viruses between 50 and 1,600CCID50/50μL. Given the cell growth condition, 100CCID50/50μL was chosen as the amount for virus used in pNT.
Figure 3. Optimization of the pNT method. (a) Linear relationship between dilution and luciferase activity. A linear relationship was observed with pseudoviral titer ranging from 50 to 1,600 CCID50/50 µL. Mahoney R2 = 0.996, MEF-1 R2 = 0.997, and Saukett R2 = 0.999. It is of note that the titer in the x-axis has been converted into lg form. Each dilution depicts average values from eight wells (replicates). Given the cell growth condition, 100 CCID50/50 µL was chosen as the challenge amounts of pseudovirus in pNT. (b) RLU-time curves of 100CCID50 pseudo-PV expressing luciferase. Increasing intensity of the fluorescent signal was observed over time, with a plateau being observed after 8h for pseudo-Mahoney and pseudo-MEF-1 respectively, while it took 14h for pseudo-Saukett to reach the plateau, with each time point depicting average values from eight wells (replicates). Therefore, 12h was chosen for the final RLU readout for all three serotypes.
As expected, luciferase activities in pseudovirus-infected RD cells were found to increase over time. As shown in , RLU values for pseudoviruses Mahoney and MEF-1 reached to the plateau at 8h, while it took 14h for pseudovirus Saukett achieving the same level, with the signal after that time remaining relatively stable or declining slightly (). Therefore, we chose 12h of infection as the final RLU readout for all three viruses and established our final pseudovirus neutralization test procedures.
The reproducibility of pNT
Serum samples with the high, medium, and low titers were used to evaluate the reproducibility of established pNT. Each sample was tested six times (). In our results, all serum titer changes were found to be within a two-fold range for 3 types and the CV values were 3.31–6.20% for pseudo-Mahoney, 6.20–8.81% for pseudo-MEF-1 and 3.03–7.00% for pseudo-Saukett, respectively, indicating the high reproducibility of the neutralization test based on pseudoviruses.
Table 1. Primers for the construction of pseudovirus.
Table 2. Reproducibility of pNT (log2).
Comparison between pnt and cNT
To determine the correlation between pNT and conventional neutralization assay cNT, 120 post-vaccination serum samples from phase II clinical trials of sIPV vaccines were selected by taking into the account of the distribution of antibody titers within each group (see section 2.1). We compared the pNT results with those generated with cNT,in which wt viruses were used,by spearman correlation statistical analysis. As shown in , there is a statistically significant strong correlation between pNT and cNT as demonstrated by coefficients for each viral strain, with r = 0.8989 (0.8563, 0.9294), p < 0.0001 for Mahoney; r = 0.8533 (0.7938, 0.8966), p < 0.0001 for MEF-1 and r = 0.8013 (0.7238,0.8588), p < 0.0001 for Saukett.
Figure 4. The agreement analysis between pNT and cNT. The pNT results of 120 post-vaccination serum samples from phase II clinical trials of sIPV vaccines were compared with those generated with cNT in which wt viruses were used. Spearman correlation statistical analysis showed it is statistically significant between pNT and cNT (p < 0.0001) for three serotypes respectively, with coefficients r = 0.8989 (0.8563, 0.9294) for Mahoney, r = 0.8533 (0.7938, 0.8966) for MEF-1 and r = 0.8013 (0.7238, 0.8588) for Saukett.
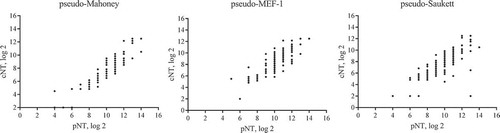
Similar values of Geometric Mean Titer (GMT) distribution across different immunization groups was found between pNT and cNT (). As expected, GMT values in wIPV serum group were higher than those obtained from sIPV and OPV groups, given antibodies from wIPV vaccine group were matched with the wt antigens, whereas sIPV and OPV vaccines were derived from Sabin strain which should be different from the test antigen. It is of note that regardless of the method used, the results from the sIPV group were lower than those from the other two groups. Since the sIPV group was sera from humans immunized with high, middle, and low doses, the data presented for this group serves only for the purpose of comparison of testing methods and does not represent true values of immunogenicity of this group.
Table 3. The geometric mean titers(GMTs) in different groups tested by pNT and cNT (log2).
Interestingly, the results of pNT from different immunization groups were significantly higher than those of cNT by about four-fold,suggesting that the pNT method presented here may be more sensitive than traditional methods.
Discussion
According to requirements outlined in ref.8, traditional wIPV vaccines should be used as reference vaccines in sIPV vaccine clinical trials.Citation8 As a result, separate neutralization tests should be conducted using Sabin strains and wild strains for the evaluation of immunogenicity since the use of two strains would allow evaluation of vaccine-induced cross-reactivity. According to GAPIII requirements, neutralization tests with wild strains must be tightly regulated and conducted in level 3 biocontainment laboratories (BSL-3).Citation10,Citation13,Citation18 In this study, we constructed three types of pseudoviruses to replace the wild poliovirus strains in the neutralization method, making it unnecessary to conduct the neutralizing assay at the BLS-3 laboratory.
Liu et al. have developed neutralization assay based on pseudovirus originated from Sabin strains with replicons based on the same originCitation19, while Arita et al. developed three serotypes of pseudovirus based on wild-strain, which involved a replicon based on Mahoney virus that has a firefly luciferase gene instead of the capsid coding region.Citation20 Notably, as the Sabin strain is safer to use, we use the functional gene of the attenuated Sabin2 strain to build a replicon for the Mahoney, MEF-1 and Saukett pseudovirus clones. We found that the structure of the pseudovirus produced by the capsid protein of different wild strains packaging the replicon is correct and the titer meets the experimental requirements. As such, employing the same replicon facilitated the speedier establishment of different type pNT experiment. Different criteria for endpoint calculation were reported when pNT assay was used for the determination of antibody titer. In most reports, RLU threshold values are set as the decrease in RLU by half.Citation21-Citation24 Similar criterion was used in this study because results from our three pseudoviruses were highly correlated with those of cNT method, with all correlation coefficients greater than 0.8. In our study, GMT detected using pNT method was approximately four times higher than that detected using cNT method. In addition, our laboratory also constructed Sabin2-based pseudovirus and observed the similar trend (unpublished data). Collectively, these results suggest that pNT established in this report is more sensitive than traditional neutralization assays and could be accurate for the analysis of weakly positive antisera. For example, a sensitive neutralizing assay would be better suited for potency test of sIPV vaccines currently conducted in rats, which usually produce weak antibody responses following immunization of type II of sIPV vaccine (data not shown).
It is of note that the sample size can be increased in order to further validate the pNT assay, which can be addressed in future studies. In summary, we have developed a pseudovirus-based neutralization assay without the use of the wt virus. It takes only one day to complete the assay whereas it takes 5–7 days to complete the traditional assay using the wt virus. This safe and sensitive assay could be a viable alternative approach to facilitate the development of polio vaccines.
Materials and methods
Cells, viruses, antibodies, and human serum samples
Human rhabdomyosarcoma(RD) cells and 293FT cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, code no.10100147) and 100U/mL penicillin-streptomycin (Gibco, code no.10378016) at 37°C and 4% CO2. The attenuated Sabin 2 poliovirus strain (GenBank accession number AY184220.1) was maintained in our lab. The wild capsid P1 cDNA sequences (Mahoney: GenBank accession no. V01149.1; MEF-1: GenBank accession no. AY238473.1; Saukett: GenBank accession no. L23847.1) were synthesized by Invitrogen Corporation (Beijing, China), while the pcDNA6.0 plasmid was also obtained from Invitrogen. Human serum samples from phase II clinical trials of sIPV vaccines were kindly provided by the Institute of Medical Biology, the Chinese Academy of Medical Sciences. Of the serum samples used in this study, samples #35, #37, and #47 are obtained from the immunization group of tOPV, wIPV, and sIPV, respectively.
Construction of capsid protein expression vectors for mahoney, MEF-1, and Saukett viruses
Construction of pseudoviruses was conducted as described by Arita et al.,Citation20 with some modifications for capsid protein expression and replicon construction. In brief, the gene of capsid proteins for wild-type strain poliovirus Mahoney, MEF-1, and Saukett were fused to EGFP by using primers FEGFP+P1-Mahoney/REGFP+P1-Mahoney, FEGFP+P1-MEF/REGFP+P1-MEF, and FEGFP+P1-Saukett/REGFP+P1-Saukett (see ) to facilitate the detection of P1 protein expression. We also added cleavage sites of 2Apro protease between the two sequences (). As the expressed P1 proteins were cleaved into VP1-VP4 capsid proteins, the EGFP was also removed for appropriate pseudoviral packaging.
As the target gene sequences carrying the restriction sites of pcDNA6.0 vector (Invitrogen, code no.V22220), the in-fusion cloning methods were adopted to construct the expression vector.Citation25 For this purpose, primers FEGFP+P1-Mahoney-6.0/REGFP+P1-Mahoney-6.0, FEGFP+P1-MEF-6.0/REGFP+P1-MEF-6.0, and FEGFP+P1-Saukett-6.0/REGFP+P1-Saukett-6.0 (see ) were designed to ligate the respective P1-EGFP fusion proteins with pcDNA6.0 vectors. The final recombinant pcDNA6.0-P1-EGFP plasmids were verified through DNA sequencing and subsequently used to transfect 293FT cells to express the capsid proteins for pseudovirus packaging.
Construction of RNA replicon
Unlike in previous studies, all pseudoviral packaging replicons used in this study were from the Sabin2 viral genome. For the purpose, the P1 fragment of Sabin2 genome was replaced by the luciferase gene, with the insertion of a 2Apro restriction site between luciferase and P2 fragments (). In brief, Sabin2 viral genomic RNA was extracted and transcribed into cDNA; the sequences between 5ʹUTR and 3ʹtermini, i.e. from P2 to the end of the genome, were amplified through PCR and ligated into the pMD19T cloning vector, respectively (Takara, code no.D102A). Two plasmids were used as the templates for the following construction. The 5ʹUTR and luciferase gene were fused by PCR using primers F5ʹUTROL/R5ʹUTROL and FluciferaseOL/RluciferaseOL. Similarly, F5ʹUTR+luciferase+3ʹend and R5ʹUTR+luciferase+3ʹend primers were used to create a 5ʹUTR+ luciferase+ 3ʹend fusion fragment. This fragment was cloned into pGEM-T Easy Vector, which has T7 promotor for transcription in vitro, with primers Freplicon and Rreplicon-polyA, in which a poly(A) tail was introduced at the 3ʹ-terminus of the fusion product to ensure its structural stability.Citation26 The primers used are also shown in .
The constructed replicon DNA vector was linearized by SalI restriction enzyme digestion (NEB, code no.R3138); replicon RNA was obtained in vitro using RiboMAX large-scale RNA production Kit (Promega, code no.P1300) (). The resultant replicon RNA was purified by isopropanol precipitation and stored at −70°C for subsequent transfection.
Pseudoviruses preparation
Pseudoviral packaging was performed through sequential transfections of capsid protein expression vectors and replicon RNA into 293FT cells (). In brief, 293FT cells were digested and plated in six-well plates at a density of 105 cells/well and incubated at 37°C for 12h. 3μg pcDNA6.0-P1-EGFP vector was transfected according to the manual of lipo3000 transfection reagent (Invitrogen, code no.L3000015). The expression of fusion proteins was monitored under a fluorescence microscope 24h after transfection, followed by transfection of 3μg replicon RNA. 24h later, cell cultures were subjected to two freeze/thaw cycle and subsequently harvested. Cellular debris was removed by centrifugation at 1700 × g for 5 min, with the supernatant filtered using a 0.22μm membrane (PALL, code no.4612) and eventually stored at −70°C prior to use.
Identification of pseudoviruses
The harvested pseudovirus supernatant was loaded onto 10% Tris-glycine gels and separated by SDS-PAGE. The protein bands were then transferred onto a nitrocellulose membrane (Millipore, code no.HATF00010) which was subsequently blocked overnight at 4°C in PBST buffer containing 0.05% Tween-20 (Sigma, code no.P1379) and 5% skim milk (BD, code no.212300). The membranes were incubated with polyclonal rabbit anti-Mahoney or MEF-1 or Saukett antibodies at 25°C for 2h. After washing with PBST 3 times, the membrane was incubated with horseradish peroxidase(HRP)-conjugated anti-rabbit IgG (Sigma, code no.A6154) at 25°C for 1h. Then the membrane was washed with PBST 5 times, added the enhanced chemiluminescence (ECL) reagent (GE, item no. RPN2232) as the substrate, exposed for 1min and visualized with chemiluminescence imaging (Chemi Doc-it HR 410 Imaging System, UVP, America).
To further confirm the type specificity, the pseudoviruses were used to measure the neutralization titer of mouse sera, which were prepared by immunization with each type of vaccine bulks. Briefly, serum samples were first inactivated at 56°C for 30min and then serially diluted by two-fold in a 96-well fluorescence detection plate. Equal volumes(50μL) of pseudovirus containing 100CCID50 were added and then incubated for 2h at 37°C, followed by the addition of 100μL RD cell suspension at a density of 3 × 104cells/well. The supernatants were discarded following 12h incubation at 37°C and 100μL of a pre-mixed chromogenic substrate (containing cell lysis buffer and luciferase substrate at a ratio of 1:1) was added to each well. The plates were briefly shaken to mix and placed at room temperature for 2 min before the RLU detection. Then the pseudovirus infection rates at different dilution point were calculated and compared.
Detection of pseudovirions using transmission electron microscopy
A total of 10 mL sample was collected from each pseudovirus type and ultra-centrifuged (Optima L-100 XP, Beckman Coulter, America) at 4°C and 40,000 × g for 2 h. The supernatant was discarded whereas the pellet was resuspended in 50μL PBS. 10μL samples were added to a copper mesh. After fixing in 1% osmium tetroxide, pseudovirions were observed under a transmission electron microscope (JEM-1220, JEOL Datum, America) at an accelerating voltage of 80kV and a magnification of 300,000 × .
Determination of pseudovirus titers
The titration of pseudoviruses was performed as described.Citation27 Pseudoviruses collected were serially diluted by five-fold from columns 1 to 11 in a 96-well plate (Costar, code no.3585) (eight wells per dilution, 100μL/well). Digested RD cells were then added at a density of 3 × 104cells/100μL for each well, mixed, and incubated at 37°C for 12h. A total of the 100μL supernatant was discarded from each well and 100μL Bright-Glo Luciferase Assay reagents (Promega, code no.E2650) were added to each well. The plates were left in darkness for 2 min at room temperature; afterward, the 150μL mixture was transferred to a 96-well test plate (Costar, item no. 3610) for chemiluminescence detection (GloMax® Navigator, Promega, America). The RLU three times higher than those of the blank controls was considered to be positive, with virus titers determined using the Spearman-Karber method.
Optimization of pseudoviral infection dose and time
We used 50–1,600CCID50/50μL of pseudoviruses to infect RD cells(3 × 104cells/well) seeded 12h earlier in the 96-well plates. After an additional incubation for 12h, RLUs(on a lg scale) were determined and plotted against their challenging amounts, while the titer value of different pseudovirus amounts was converted into lg form in order to better observe the linear relationship. The final challenging amount of pseudovirus for neutralization test (pNT) was chosen from those demonstrating a linear regression.
For time point determination, pseudoviruses were added at a volume of 50μL/well to the RD cell(3 × 104cells/well) and incubated for 2-16h. The time at which RLU values reached the plateau was taken for final neutralization test. Finally, pseudoviral neutralization test procedures were optimized based on the amount of pseudovirus and time for RLU.
Pseudovirus based neutralization test (pNT)
Serum samples were first inactivated at 56°C for 30min and then diluted by eight-fold. For the neutralization test, they were serially diluted by two-fold in a 96-well fluorescence detection plate. Afterward, equal volumes(50μL) of pseudovirus containing 100CCID50 were added and then incubated for 2h at 37°C, followed by the addition of 100μL RD cell suspension at a density of 3 × 104cells/well. Each 96-well plate contained eight wells of cells and pseudovirus controls, respectively. The supernatants were discarded following 12h incubation at 37°C and 100μL of a pre-mixed chromogenic substrate (containing cell lysis buffer and luciferase substrate at a ratio of 1:1) was added to each well. The plates were briefly shaken to mix and placed at room temperature for 2 min before the RLU detection. The pseudovirus infection rates were calculated for each dilution (infection rate = [RLUserum − RLUcellcontrol]/[RLUpseudoviral control− RLUcell control] × 100%), where the highest serum dilution showing infection rates of less than 50% was determined as the neutralizing antibody titers. As additional controls, a viral back titration control and a serum quality control were also included in each run of the test.
To assess the reproducibility of our pseudoviral neutralization tests, we selected sera with high, medium, and low titers in the assay development, with each sample tested six times. The reproducibility of pNT assay was evaluated by calculating the coefficient of variation using Graph Pad Prism 5.0.
Determination of correlation between pNT and cNT
The results of cNT were obtained from the CDC laboratory in the United States, with neutralizing antibody titers against Mahoney, MEF-1, and Saukett strains were determined. Neutralizing antibody testing was performed using the WHO standard protocol.Citation27-Citation29 Briefly, diluted serum was mixed with an equal volume of medium(50μL) containing 100CCID50 of poliovirus in 96-well plates and incubated for 2h at 37°C, followed by the addition of 100μL of Hep-2 cell suspension (2 × 104 cells). After incubation for 5 days at 36°C, the cytopathic effect of viral infection was examined. The serum antibody titer is the highest dilution of serum that protects 50% of the culture against 100CCID50 of a viral challenge using the Reed-Muench method. Results generated with pNT were compared with those obtained with cNT, with spearman correlation analysis performed using GraphPad Prism5.0 software.
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the authors.
Acknowledgments
We would like to express sincere gratitude to the Institute of Medical Biology, Chinese Academy of Medical Sciences for providing us with the samples. We would also like to thank the US CDC for providing us with serum samples and sharing the antibody test results.
Additional information
Funding
References
- World Health Organization. Polio vaccines: WHO position paper, January 2014–recommendation. Vaccine. 2014 Jul 16;32(33):4117–4118. doi:10.1016/j.vaccine.2014.04.023.
- Plotkin SA, Orenstein W, Offit PA. Vaccines,Fifth Edition. Singapore: Elsevier Saunders; 2011
- Platt LR, Estívariz CF, Sutter RW. Vaccine-associated paralytic poliomyelitis: a review of the epidemiology and estimation of the global burden. J Infect Dis. 2014 Nov 1;210(suppl 1):S380–9. doi:10.1093/infdis/jiu184.
- Global Polio Eradication Initiative. Polio today. [accessed 20 Jul 2018]. http://polioeradication.org/polio-today/polio-now/thisweek.
- Foiadelli, T, Savasta S, Battistone A, Kota M, Passera C, Fiore S, et al. Nucleotide variation in Sabin type 3 poliovirus from an Albanian infant with agammaglobulinemia and vaccine-associated poliomyelitis. BMC Infectious Diseases, 16 (1),277. doi:10.1186/s12879-016-1587-y.
- Kew OM, Sutter RW, de Gourville EM, Dowdle WR, Pallansch MA. Vaccine-derived polioviruses and the endgame strategy for global polio eradication. Annu Rev Microbiol. 2005;59(1):587–635. doi:10.1146/annurev.micro.58.030603.123625.
- Minor P. Vaccine-derived poliovirus (VDPV): impact on poliomyelitis eradication. Vaccine. 2009 May 5;27(20):2649–2652. doi:10.1016/j.vaccine.2009.02.071.
- World Health Organization. Recommendations to assure the quality, safety and efficacy of poliomyelitis vaccine (inactivated). Technical Report SeriesNo.993. Geneva (Switzerland): WHO; 2015. [accessed 08 Feb 2018]. http://www.who.int/biologicals/vaccines/Annex3_IPV_Recommendations_eng.pdf.
- World Health Organization. Meeting of the Strategic Advisory Group of Experts on immunization, April 2013 – conclusions and recommendations. Weekly Epidemiol Record. 2013;88(20):201–206.
- World Health Organization. Initiative GPE polio eradication & endgame strategic plan 2013–2018. Immunization Programs; 2013 [accessed 09 Feb 2018]. http://polioeradication.org/wp-content/uploads/2016/07/PEESP_EN_A4.pdf.
- Dowdle W, van der Avoort H, de Gourville E, Delpeyroux F, Desphande J, Hovi T, Martin J, Pallansch M, Kew O, Wolff C. Containment of polioviruses after eradication and OPV cessation: characterizing risks to improve management. Risk Anal. 2006;26(6):1449–1469. doi:10.1111/j.1539-6924.2006.00844.x.
- National Board of Health and Family Planning Commission for Disease Control and Prevention. China responds to WHO resolution on new immunization strategy for polio vaccine; 2016 [assessed 2018 Feb 08]. http://www.nhfpc.gov.cn/jkj/s3582/201604/8c760a934d5b4d41a81752915c58d304.shtml.
- World Health Organization. Global Action Plan to minimize poliovirus facility-associated risk after type-specific eradication of wild polioviruses and sequential cessation of oral polio vaccine use. WHO/POLIO; 2015 May 15 [assessed 2018 Feb 08]. http://polioeradication.org/wp-content/uploads/2016/12/GAPIII_2014.pdf.
- Fukushi S, Mizutani TM, Kurane I, Taguchi F, Tashiro M, Morikawa S. Evaluation of a novel vesicular stomatitis virus pseudotype-based assay for detection of neutralizing antibody responses to SARS-CoV. J Med Virol. 2006;78(12):1509–1512. doi:10.1002/jmv.20732.
- Qiu C, Huang Y, Zhang A, Tian D, Wan Y, Zhang X, Zhang W, Zhang Z, Yuan Z, Hu Y, et al. Safe Pseudovirus-based assay for neutralization antibodies against Influenza A(H7N9) virus. Emerg Infect Dis. 2013;19(10):1685–1687. doi:10.3201/eid1910.130728.
- Zhao G, Du L, Ma C, Ye L, Lin L, Poon VK, Wang L, Fei Y, Zheng BJ, Jiang S, et al. A safe and convenient pseudovirus-based inhibition assay to detect neutralizing antibodies and screen for viral entry inhibitors against the novel human coronavirus MERS-CoV. Virol J. 2013;10:266. doi:10.1186/1743-422X-10-266.
- Martín J. Poliovirus. New York (NY): Springer; 2016.
- World Health Organization. Dept. of immunization, vaccines and biologicals. Polio laboratory manual. 4th ed. World Health Organization: Geneva,Switzerland. WHO/IVB; 2004 Oct 04 [assessed 2018 Feb 08]. http://who.int/iris/handle/10665/68762.
- Liu S, Song D, Bai H, Lu W, Dai X, Hao C, Zhang Z, Guo H, Zhang Y, Li X. A safe and reliable neutralization assay based on pseudovirus to measure neutralizing antibody titer against poliovirus. J Med Virol. 2017 Dec;89(12):2075–2083. Epub 2017 Aug 29. doi:10.1002/jmv.24909.
- Arita M, Iwai M, Wakita T, Shimizu H. Development of a poliovirus neutralization test with poliovirus pseudovirus for measurement of neutralizing antibody titer in Human Serum. Clin Vaccine Immunol. 2011;18(11):1889–1894. doi:10.1128/CVI.05225-11.
- Buck CB, Pastrana DV, Lowy DR, Schiller JT. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol Med. 2005;119:445–462. doi:10.1385/1-59259-982-6:445.
- Tamin A, Harcourt BH, Lo MK, Roth JA, Wolf MC, Lee BH, Weingartl H, Audonnet JC, Bellini WJ, Rota PA. Development of a neutralization assay for Nipah virus using pseudotype particles. J Virol Methods. 2009;160(1–2):1–6. doi:10.1016/j.jviromet.2009.02.025.
- Arya SC, Agarwal N. Poliovirus neutralization test with poliovirus pseudovirus to measure neutralizing antibody in humans. Clin Vaccine Immunol. 2012;19(3):458;author reply 459. doi:10.1128/CVI.05568-11.
- Mel K, Darrel C, Amanda Y, Ron C, Qiang S, Wendy M, Shelly MN, Deborah M, Marc D, Joel P, et al. Assessment of HPV 16 and HPV 18 antibody responses by pseudovirus neutralization, Merck cLIA and Merck total IgG LIA immunoassays in a reduced dosage quadrivalent HPV vaccine trial. Vaccine. 2014;32(5):624–630. doi:10.1016/j.vaccine.2013.09.007.
- Irwin CR, Farmer A, Willer DO, Evans DH. In-Fusion® cloning with vaccinia virus DNA polymerase. Methods Mol Biol. 2012;890:23–35. doi:10.1007/978-1-61779-876-4_2.
- Koch F, Koch G. The molecular biology of poliovirus. Vienna: Springer-Vienna; 1985. doi:10.1007/978-3-7091-7000-7
- WHO Expanded Programme on Immunization World Health Organization. Division of communicable diseases. Manual for the virological investigation of poliomyelitis; 1990 [assessed 2018 Feb 08]. http://apps.who.int/iris/handle/10665/62186.
- Liao G, Li Q. Safety and immunogenicity of inactivated poliovirus vaccine made from Sabin strains: a phase II, randomized, positive-controlled trial. J Infect Dis. 2012;205(2):237–243. doi:10.1093/infdis/jir723.
- Sun M, Li C, Xu W, Liao G, Li R, Zhou J, Li Y, Cai W, Yan D, Che Y, et al. Immune serum from Sabin inactivated poliovirus vaccine immunization neutralizes multiple individual wild and vaccine-derived polioviruses. Clin Infect Dis. 2017;64(10):1317–1325. doi:10.1093/cid/cix110.