
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
In chronic Hepatitis B Virus (HBV) infections HBV-specific T cells are functionally impaired. Immunotherapy may restore HBV-specific T cell responses essential for sustained disease remission off-treatment and induction of a functional cure. Chimigen® Molecules are fusion proteins of antigen(s) with the Fc fragment of a xenotypic antibody designed to target specific receptors on dendritic cells (DCs). Here we describe the production and pre-clinical evaluation of Chimigen® HBV (C-HBV), containing HBV PreS1 and PreS2 peptide fragments, HBV core and murine Fc, produced in insect cells. C-HBV binding to immature DCs and internalization by endocytosis was FcγRII (CD32) and mannose receptor (CD206) dependent and led to increased MHC I and MHC II surface expression. Upon exposure of human T cells isolated from HBV un-infected healthy and chronically HBV-infected donors to C-HBV-pulsed mature DCs ex vivo, C-HBV induced vigorous T cell proliferation and enhanced expression of IFN-γ, TNF-α, perforin and granzyme B in both CD4+ and CD8+ T cell subsets. Re-stimulation of C-HBV-activated T cells from chronically infected donors with HBV PreS1/PreS2 and core overlapping peptides induced IFN-γ production in both CD4+ and CD8+ populations. C-HBV-activation of peripheral blood mononuclear cells (PBMCs) from chronically HBV-infected patients stimulated granzyme B production by CD4+CD25− T responder (Tresp) cells, accompanied by an increase in Annexin V staining on CD4+CD25+ T regulatory (Treg) cell phenotype, consistent with apoptosis. The observed HBV-specific cellular responses induced by C-HBV ex vivo suggest that C-HBV is a promising immunotherapeutic candidate for the treatment of chronic HBV infections.
Introduction
More than 2 billion people worldwide have been infected by the hepatitis B virus (HBV), and among this population, an estimated 257 million are chronically infected, thus hepatitis B remains a major health concern.Citation1 Many patients with chronic hepatitis B infection are at a high risk for developing liver cirrhosis and hepatocellular carcinoma. Worldwide, an estimated 887,000 people died due to complications resulting from chronic HBV infection in 2015.Citation1
Prophylactic vaccines containing the small HBV surface antigen (HBsAg) are effective in eliciting protective immunity against HBV infection,Citation2 but do not promote resolution of established chronic infections. HBsAg-based vaccines produce neutralizing antibodies which block HBV entry into uninfected hepatocytes. However, these are ineffective for established chronic/persistent HBV infections. There is considerable evidence from various studies that in the resolution of HBV infection, HBV-specific T cells are the major players and among them core-specific T cells are the most important.Citation3–Citation5 An effective HBV immunotherapy should be able to induce T cell activation which has been lacking, for various reasons. New antibodies produced may also play a role in assisting the cellular response to clear the virus and viral antigens. To date, treatment options for patients with chronic hepatitis B consist mainly of pegylated interferon-α or nucleoside/nucleotide analogues such as entecavir and tenofovir. Current therapies achieve sustained off-treatment responses in a minority of highly selected patients, and a functional cure, hallmarked by a sustained clearance of circulating HBsAg and normalization of aminotransferase (ALT) levels following discontinuation of treatment remains most difficult to achieve.Citation6 Hence, the emergence of drug-resistant strains of virus during long-term suppressive therapy with nucleoside/nucleotide analogues and cost remain as major concerns.Citation7 Unlike hepatitis C virus infections,Citation8 a virological cure for chronic HBV might not be possible due to the stability of covalently closed circular (ccc) HBV DNA and the lack of a cellular mechanism for its elimination.Citation9
Functional impairment of HBV-specific immune response is a key feature of chronic HBV infectionCitation10,Citation11 and has been attributed to many factors including T cell exhaustionCitation12 associated with overexpression of inhibitor receptors such as programmed cell death-1 (PD-1) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) on T cells.Citation13 Fisicaro et al.Citation14 have suggested mitochondrial dysfunction as a cause of T cell exhaustion in HBV-infected individuals. Another proposed mechanism for the impaired T cell response is the increasing number of regulatory T cells (Treg) during chronic infection which suppress or downregulate the induction and proliferation of effector T cellsCitation15 and dendritic cell dysfunction.Citation16 Immunotherapeutic approaches which aim to induce/restore effective cellular and humoral responses against HBV antigens have been of considerable interest in recent yearsCitation17,Citation18 but have not been shown to be able to elicit clinically relevant responses to date.Citation19,Citation20 An ideal immunotherapy would “break” the immune tolerance status in chronic HBV-infected hosts to restore specific antiviral immunity and eliminate persistent infection by activating T cell responses which appear to be critical in achieving a functional cure.
Based on the observation that spontaneous resolution of HBV is accompanied by immune system re-constitution,Citation21 various immunotherapeutic approaches to induce novel HBV-specific cellular and humoral responses have been attempted.Citation22–Citation24 These immunotherapy agents include DNA and viral vectors, proteins or peptide-based vaccines with various adjuvants. Many of these approaches so far have not been successful.
Chimigen® molecules are a novel class of immunotherapy agents that target viral antigens to antigen presenting cells, especially dendritic cell (DC) receptors, for efficient uptake, processing, and presentation via major histocompatibility complex class I (MHC I) and class II (MHC II) pathways. DCs play a central role in the activation of the immune responses.Citation25 Antigen uptake by DCs is mediated by receptors, such as C-type mannoseCitation26 and Fcγ receptors.Citation27 A major advantage of targeting receptors on DCs is the facilitation of antigen delivery to appropriate compartments for the induction of an effective immune response without the use of adjuvantCitation28,Citation29 and with reduced antigen dose.Citation30
Chimigen® HBV (C-HBV) consists of an immune response domain (IRD) containing HBV PreS1 peptide fragment (119 amino acids), PreS2 peptide fragment (55 amino acids), the entire core antigen (185 amino acids), and a target binding domain (TBD) comprised of a linker fragment of eight amino acids and xenotypic Fc fragment from mouse IgG1 (). The order of the antigens was similar to the open reading frame of the HBV genome. The TBD mediates the binding to specific DC receptors such as Fcγ receptors.Citation27 The xenotypic nature of the murine Fc makes the molecule more immunogenic. In addition, the expression of the fusion protein in Sf9 insect cells which imparts primitive non-mammalian glycosylation without terminal sialic acid residues,Citation31,Citation32 increases the immunogenicityCitation33 of the molecule and also permits the targeting of mannose receptors (CD206) for antigen uptake, processing, and presentation.Citation34
Figure 1. Schematic representation of C-HBV.
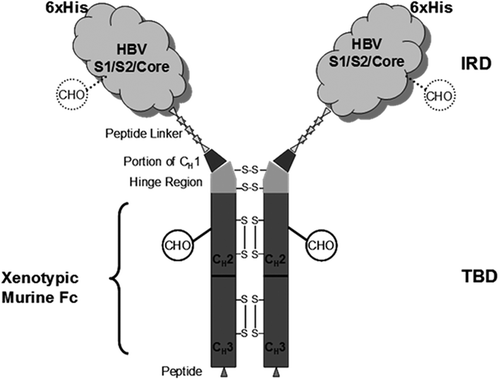
The rationale for including HBV Core and PreS1/Pre2 antigens as components of C-HBV was based on prior studies showing the efficacy of these antigens in eliciting T cell immunity. The effect of circulating viral HBeAg and HBsAg as well as the lack of sufficient T cell responses to HBV core are two of several mechanisms suggested to explain the establishment of chronicity by HBV.Citation35 Resolution of chronic HBV achieved by adoptive transfer of bone marrow from HLA-identical donors with natural immunity to HBV correlated with core-specific T cell activation,Citation3 and several strategies have been attempted to improve T cell responses to core.Citation36–Citation41 It has been suggested that PreS1/S2 peptides may also be relevant for eliciting immune responses.Citation38,Citation42,Citation43 PreS protein was shown to be capable of stimulating preS-specific CD8+ and CD4 + T cell responses in BALB/c mice and HBV transgenic mice. Furthermore, preS immunization provided protection against hydrodynamic transfection of HBV DNA in mice.Citation42 These studies suggest that preS protein may also elicit cellular responses against HBV.
Here we describe the production of Chimigen® HBV (C-HBV) in insect cells and the evaluation of its ability to elicit HBV-specific T cell responses ex vivo, with cells derived from uninfected healthy and chronically HBV-infected donors.
Materials and methods
Cloning of C-HBV
DNA sequences for the HBV S1 fragment (119 amino acids), S2 fragment (55 amino acids) and entire core protein (185 amino acids) were obtained from Genbank DNA databanks (HBV 991, subtype adw, Accession Number X51970.1). The TBD sequence was derived from a murine IgG1 antibody heavy chain sequence (Genbank Accession Number D78344.1). A gene that encoded the three HBV proteins, an eight amino acid linker peptide and TBD in a single open reading frame was commercially synthesized and codon-optimized for enhanced production in insect cells by GenScript. The synthetic gene was cloned into a pUC57 plasmid and was designed to have a unique Sal I site at the 5ʹ end and a unique Hind III site at the 3ʹ end. The coding sequence of the TBD insert was modified to remove putative protease cleavage sites within the hinge region. The three mutations corresponded to R8A, K13A, and T18A (the numbering of position 1 of the TBD insert refers to the valine residue in the VDKK CH1 domain). For proper post-translational processing of the protein, the signal sequence from Autographa californica nuclear polyhedrosis virus (AcNPV) gp64 protein was cloned into pFastBac-HTa (Thermo Fisher Scientific, 10584–027). Two oligonucleotides that encode a unique 5ʹ Ava II site and a 3ʹ Rsr II site (5ʹGCATGGTCCATGGTAAGCGCTATTGTTTTATATGTGCTTTTGGCGGCGGCGGCGCATTCTGCCTTTGCGGATCTGCAGGTACGGTCCGATGC-3ʹ and 5ʹ-GCATCGGACCGTACCTGCAGATCCGCAAAGGCAGAATGCGCCGCCGCCGCCAAAAGCACATATAAAACAATAGCGCTTACCATGGACCATGC-3ʹ) were synthesized and annealed together. After digestion with Ava II (New England Biolab, R0153S), and Rsr II (New England Biolabs, R051S), the fragment was cloned into Rsr II digested pFastBac-HTa, which places the gp64 signal sequence just upstream of the 6xHis tag to generate pFastBacHTa-gp64. The S1/S2/Core/TBD insert in pUC57 was isolated by digestion with Sal I (New England Biolabs, R3138S) and Hind III (New England Biolabs, R0104S) and cloned into Sal I and Hind III digested pFastBacHTa-gp64.
Generation of baculovirus
Recombinant bacmids were generated using the Bac-To-Bac® cloning system (Thermo Fisher Scientific, 10359–016) in E. coli strain DH10Bac (Thermo Fisher Scientific,10361–012). The gene for C-HBV cloned into pFastBacHTa-gp64 was transformed into E. coli strain DH10Bac. The recombinant bacmids were isolated and used for transfecting Sf9 insect cells to generate the recombinant baculoviruses that express the recombinant protein in insect cells. The baculovirus stock was amplified to produce a high titer stock, and titer (pfu, plaque forming units per mL) was determined with the Baculovirus Titering Kit (Expression Systems, 97–101).
Production of recombinant proteins in wave bag bioreactors
Sf9 insect cells (Thermo Fisher Scientific, 11496015) were seeded at 1 × 106 cells/mL into 100 mL ESF 921 (Expression Systems, 96-001-01) media in a 500 mL flask. Cultures were incubated at 27.5ºC with shaking at 130 rpm on an Innova Model 2100 Benchtop Platform Shaker (Eppendorf, M11940000) for 3–4 d (until the cell density reached 6–8 × 106 cells/mL). When the culture reached the desired cell density, an aliquot of the culture (1 × 106 cells/mL) was seeded into 1 L ESF 921 media in a 2 L flask. Cultures were incubated at 27.5ºC with shaking at 130 rpm, in a bench-top shaker-incubator until the cell density reached 6–8 × 106 cells/mL (3–4 d).
A Wave Bioreactor System 2/10EH (GE Healthcare, 28-4115-00) and Cellbag 10L/O (GE Healthcare, CB0010L-01) was used for 5 L cultures with the ESF921 media. The seed culture (1 L), grown as described above, was used to inoculate 4 L of ESF 921 media. The rocking of the Wave Bag Bioreactor was set at 32 rpm, 5º rocking angle, atmospheric air flow at 0.30 Lpm (liters per minute) and temperature at 27.5°C. Rocking of the bag continued until the cell density reached 2–3 × 106 cells/mL.
For the production of C-HBV protein, the Sf9 cells were infected with an MOI of 2 pfu/mL. The bioreactor was allowed to rock at 32 rpm, 5º rocking angle, air flow (30% O2) of 0.30 Lpm and at 27.5ºC. The cells were harvested at 42 h following the infection by centrifugation at 1600 × g for 10 min, at 4ºC. Pellets of infected Sf9 cells were washed, frozen in liquid nitrogen and stored at −80ºC.
Purification of C-HBV
C-HBV-containing N-terminal 6xHis tag was purified using Ni-affinity chromatography. Frozen-infected Sf9 cell pellets were re-suspended in 32 mL lysis buffer (6 M guanidine HCl, 20 mM sodium phosphate, 0.5 M sodium chloride, pH 7.4) per 100 mL of frozen cell pellet. The lysate was sonicated using a Misonix 3000 Ultrasonic Liquid Processor (Misonix, S-3000) three times at 100 W for 30 s on ice. Tween-20 (1%) and imidazole (20 mM) were added to the lysate, the pH was adjusted to 7.4 and stirred at room temperature for 2 h. After stirring, the lysate was filtered through a 5 μm syringe top filter (Pall Corporation, 4650) and then a 0.45 μm syringe top filter (Pall Corporation, 4654).
The protein was purified using an AKTA Explorer 100 (GE Healthcare, 18111241). The solubilized filtered lysate was loaded onto a 5 mL HisTrap FF column (GE Healthcare, 17531901). The column was washed with 10 column volumes of 6 M guanidine HCl, 20 mM sodium phosphate, 0.5 M sodium chloride, 20 mM imidazole, 0.05% Tween 20, pH 7.4 buffer and subsequently with wash buffer 1 (6 M guanidine HCl, 20 mM sodium phosphate, 10 mM ethylenediamine, 20 mM imidazole, 0.05% Tween 20, pH 7.4) and wash buffer 2 (6 M guanidine HCl, 20 mM sodium phosphate, 40 mM imidazole, 0.05% Tween 20, pH 7.4). Finally, the protein was eluted with the elution buffer (6 M guanidine HCl, 20 mM sodium phosphate, 150 mM imidazole, 0.05% Tween 20, pH 7.4) and collected in 1 mL fractions.
The fractions containing the purified protein were pooled and concentration of the protein was determined using Compat-Able protein assay preparation reagents (Thermo Fisher Scientific, 23215) and Micro BCA assay kit (Thermo Fisher Scientific, 23235). Next, 2-mercaptoethanol (Sigma-Aldrich, M3148) to 10 mM was added and incubated for 1 h at room temperature. The purified protein was transferred into a Slide-A-Lyzer cassette (Thermo Fisher Scientific, PI-66810) and dialyzed against Redox-shuffling buffer (0.5 M guanidine HCl, 0.5 M Tris, 5 mM reduced glutathione, 0.5 mM oxidized glutathione, 0.05% Tween 20, pH 7.4) overnight at 4°C followed by dialysis against 0.5 M Tris, 0.05% Tween 20, pH 7.4 for 12 h. The protein was finally dialyzed against a buffer containing 10 mM sodium phosphate, 0.15 M NaCl, 0.05% Tween 20, pH 7.4 for a minimum of 4 h.
The purified protein was sterilized by filtration through a 0.2 µm syringe-top filter (Pall Corporation, 4602). The concentration of the purified C-HBV protein was determined using the Micro BCA assay. Purified protein is stored at 4°C and is stable for over a period of at least 18 months under these conditions.
Biochemical evaluation of C-HBV
Purified C-HBV was characterized by SDS-PAGE. The protein sample was mixed with loading buffer containing the reducing agent DTT (2 mM) (Fermentas, R0891) and loaded onto a 7.5% TGX precast polyacrylamide gel (Bio-Rad, 456–1023). The gel was run at 100 V for 75 min. PageBlue (Thermo Fisher Scientific, PI 24620) was added; the gel was stained for 1 h and destained with several changes of ddH20.
The purified protein, following electrophoresis on 7.5% SDS-PAGE, was electroblotted onto Hybond ECL nitrocellulose membranes (GE healthcare, RPN303D) and used for Western detection using antibodies specific for components of the C-HBV protein. Antibody-binding was detected by chemiluminescence using the ECL kit (GE Healthcare, RPN2232). For C-HBV, four different antibodies were used: 6xHis horseradish peroxidase-conjugated mAb (Clontech, 631210), anti-mouse IgG (Fc specific) horseradish peroxidase-conjugated antibody (Sigma-Aldrich, A0168), anti-HBV core (Abcam, ab8638) and anti-PreS2 (Abcam, ab8635) mAbs.
For glycoprotein staining, the electroblotted Hybond ECL nitrocellulose membrane was incubated in Tris-buffered saline (TBS) solution (20 mM Tris, 150 mM NaCl, pH 7.5) containing 2% Tween 20 for 10 min at room temperature. After rinsing the membrane with TBS, the nitrocellulose membrane was incubated with ConA-HRP (Sigma-Aldrich, L6397) at a concentration of 1 µg/mL in TBS with 0.05% Tween 20, 1 mM MgCl2, 1 mM MnCl2 and 1 mM CaCl2 for 30 min at room temperature. The membrane was washed with TBS containing 0.05% Tween 20 and the positive bands were visualized by chemiluminescence using the ECL kit (GE Healthcare, RPN2232).
Immunological characterization of C-HBV using PBMCs isolated from healthy non-infected and chronically HBV-infected donors
Human PBMC
PBMCs were isolated from a leukapheresis preparation obtained from a healthy donor with the HLA-A2 haplotype (Biological Specialty Corporation, 213-15-04). Leukapheresis preparations were tested with the complete battery of FDA required infectious disease state tests and verified to be HBsAg negative, HBcAg antibody negative, and HBV DNA negative. PBMCs were obtained by Ficoll-Hypaque (Sigma-Aldrich, GE17-1440-02) density gradient centrifugation. PBMCs at 3 × 107 cells/cryovial (Thermo Scientific Nalgene, AY509X32) in freezing media (40% matched donor plasma, 50% AIM V (Thermo Fisher Scientific, 12055–091) and 10% DMSO), were stored in liquid nitrogen.
Chronic hepatitis B carriers that provided blood for isolation of PBMCs for this study were enrolled in study MS10 Pro00001044 approved by the Health Research Ethics Board of the University of Alberta. For this study, inactive hepatitis B carrier patients who had not received any anti-viral treatment were recruited from the outpatient clinics of the Liver Unit of the Division of Gastroenterology at the University of Alberta, Edmonton. The collection of peripheral blood (60 mL in seven ACD solution A tubes, Thermo Fisher Scientific, B364606) was performed using protocols approved by the Health Research Ethics Board at the University of Alberta. Upon patient consent, blood was obtained to screen for the expression of HLA-A2. After lysing red blood cells with ammonium chloride, blood cells were washed with D-PBS and stained with an AF 488-labeled anti-HLA-A2.01 antibody (Bio-Rad, MCA2090A488) or isotype control antibody. After staining, the cells were washed with D-PBS, fixed and acquired on a flow cytometer. HLA-A2 positive and HLA-A2 negative patients were included in the study. PBMCs and plasma were separated from whole blood by Histopaque-1077 density gradient centrifugation (Sigma-Aldrich, 10771). Plasma was heat-inactivated and used for cell culture (2.5% in AIM V) and in the medium for cryopreservation (10% DMSO, 90% plasma) of PBMCs which are not used immediately in the ex vivo assays.
Isolation and differentiation of monocytes to iDC
Cryopreserved PBMCs were used to generate iDCs in a procedure modified from Whiteside et al.Citation44 PBMCs were cultured on 100 mm tissue culture dishes (BD Falcon, 353003) for 1 h at 37°C in AIM V media with 2.5% matched plasma (heat inactivated). Following culture, non-adherent cells were removed by washing with AIM V media. The adherent cells were then cultured for 24 h in AIM-V/2.5% matched serum containing IL-4 (R&D Systems, 204–01) and GM-CSF (R&D Systems, 215-GM-010) (1000 IU/mL of each). Following culture, the iDCs were harvested, washed once with AIM V media containing 2.5% matched serum, followed by two washes with Dulbecco’s phosphate-buffered saline (Thermo Fisher Scientific, 14190–250) containing 0.1% (w/v) BSA (PBSB). The phenotype of the isolated immature DCs was assessed by labeling for various cell surface markers including CD64, CD32, CD16, CD206, CD205, CD209, HLA-ABC, HLA-DR, CD14, CD11c, CD86, CD80, CD40, CD83, CD19 and CD3.Citation45
Binding of C-HBV protein to iDCs
Binding of C-HBV to iDCs prepared from uninfected donor was evaluated by flow cytometry. All steps were performed at 4°C. Cells were incubated for 60 min in PBSB containing various concentrations of protein or the corresponding dialysis buffer (2 × 105 cells/well in 96-well v-bottom plates) (Corning Costar, 3894) in a volume of 25 µL. Protein binding was detected by incubation of the cells with rat anti-mouse IgG1-biotin (BD Biosciences, 553441) in PBSB for 20 min. The cells were washed with PBSB and then incubated with Streptavidin-PE-Cy5 (BD Biosciences, 554062) for 20 min. After washing with PBSB, cells were resuspended in PBSB containing 2% paraformaldehyde (PF) and the binding assessed by FACS. To determine the specificity of the receptors which bind C-HBV, iDCs were incubated with buffer control, isotype control mAb, anti-CD32 mAb, anti-CD206 mAb, or both anti-CD32 and anti-CD206 together for 1 h at 4°C before incubation with C-HBV for 1 h at 4°C. Bound C-HBV was detected with Penta-His AF 647 (Qiagen, 35370).
FACS acquisition and analysis
Cells were acquired with a FACSCalibur fitted with CellQuest Pro acquisition and analysis software (BD Biosciences, 342974). A gate was made on the viable cell population as determined by the forward scatter (FSC) and side scatter (SSC) profiles and ≥20,000 events were acquired. The percentage of specific positive cells was calculated as (% positive cells test sample – % positive cells control)/(100 – % positive cells of control) × 100. The relative mean fluorescent intensity (MFI) was determined as MFI of the test sample – MFI of the control sample.
Confocal fluorescence imaging
An inverted laser scanning confocal microscope (Zeiss, LSM 510) with an oil-immersion objective (NA = 1.3, 40 ×, Zeiss, 420762–9800) was used for imaging. Pacific blue, AF 488, 555, 647 were excited at 405, 488, 543 and 633 laser lines, respectively. During the image collection, the background level was set to deliver a non-zero count in each pixel and the gain was set to avoid saturating any of the pixels. This protocol would ensure that no information is lost at either end of the intensity scale hence the fluorescence intensity is quantitatively imaged as a function of position across the cell. In dual or triple labeling experiments, to minimize cross-over fluorescence, images were collected separately at each wavelength. One high-resolution image (Zoom 10–12, pixel resolution = 0.03–0.04 μm) from an average of two scans was collected from each cell and 30 to 40 images from different cells were collected from different cells. All results were an average from double or triple individual experiments (total about 90 cells) to ensure the accurate determination of mean of the cell population. Cells without labeling and cells labeled with secondary antibody only were imaged to obtain images representing the autofluorescence and nonspecific labeling, respectively. All imaging was conducted at room temperature. Images were processed using ICS and ICCS software from NIH Image (http://rsb.info.nih.gov/nih-image/) plugged in Image J program. Results were derived after background corrections which subtract the auto fluorescence and nonspecific labeling.
Analysis of C-HBV binding, internalization, and processing by confocal microscopy
The binding, internalization, and processing of the C-HBV by iDCs from uninfected donor was studied by confocal microscopy. iDCs were prepared as described above and transferred to chambered slides (MatTek, CCS-8) and incubated for an additional day. This is the optimal time for DC morphology and cell surface receptor expression. For the binding assay, all steps were performed at 4°C. First, iDCs were cooled on ice for 5 min and incubated with 3% BSA in DPBS to prevent nonspecific binding before the addition of C-HBV. Cells were incubated for 30 min in PBSB with C-HBV and rinsed three times with DPBS/0.25% BSA. Protein binding was detected by incubation of the cells with F(ab’)2 fragment of goat anti-mouse AF 488 (Thermo Fisher Scientific, A-11017) for 30 min at room temperature. Cells were fixed with 4% PF for 30 min at room temperature. Cells were mounted with SlowFade Gold anti-fade reagent (Thermo Fisher Scientific, S36936) and cover slips were sealed on before confocal imaging.
To confirm that CD32 is involved in C-HBV binding to iDCs, anti-CD32 antibody was used to block the binding. iDCs were rinsed with RPMI 1640 (Thermo Fisher Scientific, 11875093) and incubated with rabbit anti-human CD32 (Bio X Cell, BE0224-R100MG, 80 µg/mL) for 1 h at 4°C. After washing, DCs were incubated with C-HBV (2 µg/mL) at 4°C for 30 min. Cells were fixed with 4% PF for 30 min at room temperature. Binding was detected by confocal fluorescence microscopy with a donkey anti-rabbit AF 488 (Thermo Fisher Scientific, A-21206, 400 µg/mL) followed by goat anti-mouse AF 555 (Thermo Fisher Scientific, A28180, 6 µg/mL).
Internalization of C-HBV by iDCs was studied either by directly incubating the cells in media containing C-HBV at 37°C, 7% CO2 or by first labeling the surface receptors at 4°C in PBSB, washing away the unbound protein, and then studying the uptake of receptor-bound C-HBV over time (0, 10, 30, and 60 min) and at different concentrations (1, 2, 4, 10, 20, 100, 400 µg/mL) at 37°C, 7% CO2 in RPMI/2.5% matched plasma. Internalization was stopped by soaking the cells with cold PBS. Cells were fixed with 4% PF at room temperature for 30 min, permeabilized with 0.5% Triton X-100, 10% FBS, 3% BSA in PBS for 10 min and C-HBV detected with goat anti-mouse IgG1 AF 555 (Thermo Fisher Scientific, A-21127,10 µg/mL) by confocal microscopy.
Co-localization with transferrin
In order to study receptor-mediated endocytosis of C-HBV, C-HBV loaded DCs were incubated with goat anti-Fc IgG-AF 555 (Thermo Fisher Scientific, A-21127) for 3 h at 37°C/7% CO2, washed, and incubated with AF 488 transferrin conjugate (Thermo Fisher Scientific, T13342) for 1 h at 37°C/7% CO2 or incubated with both goat anti-Fc Fab IgG-AF 555 and AF 488 transferrin conjugate for 3 hr at 37°C/7% CO2. After washing, cells were fixed with 4% PF for 30 min at room temperature, mounted and analyzed by confocal microscopy.
Co-localization with MHC I and II
The expression of MHC I & II molecules and their co-localization with C-HBV were examined using confocal microscopy. iDCs were incubated in AIM V medium with donor-matched serum and cytokines (IL-4, GM-CSF) for 3 d. Following this, they were incubated in RPMI 1640 media alone (control), with LPS (100 ng/mL, Sigma-Aldrich, L4391), or with C-HBV (2 µg/mL) at 37°C for 4, 8 or 24 h, the cells were washed and fixed using 4% PF at 4°C for 30 min. Cycloheximide (5 μg/mL. Sigma-Aldrich, C4859), used to inhibit protein synthesis, was added 30 min prior to initiation of experiment and remained present in the media during incubation. The cells were permeabilized using 0.5% Triton X-100 in FBS at room temperature for 10 min. Following this, the cells were labeled with mouse anti-human HLA-DR (MHC II) Pacific Blue (BioLegend, 327016, 10 µg/mL), mouse anti-human HLA-ABC (MHC I) AF 647 (BioLegend, 311416, 10 µg/mL), mouse anti-human calnexin (ER marker) FITC (Novus Biologicals, NBO2-36570 F, 5 µg/mL), LAMP (late endosome/lysosome marker) AF 488 (BioLegend, 121607, 10 µg/mL), or goat anti-mouse IgG1 (C-HBV) AF 555 (Thermo Fisher Scientific, A-21127,10 µg/mL).
Antigen Presentation Assays (APAs) and generation of antigen-loaded mature DCs
The immune responses of naïve T cells from uninfected donor to antigen presented by DCs were assessed using APAs. The general protocol for the APA is schematically shown in . For chronic HBV-infected donor samples, due to the limitation of volume of samples, antigen was added to whole PBMCs.
Figure 2. Antigen Presentation Assay (APA) schematic.
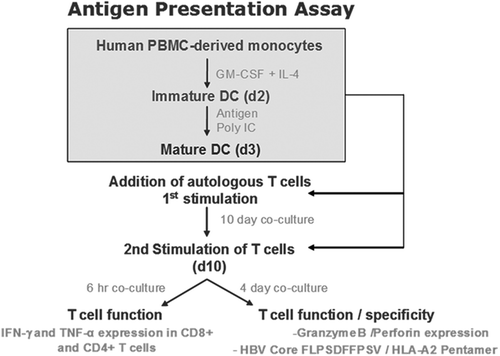
Immature DCs were generated as described above and incubated for 8 h with antigen or buffer (control). The cells were then cultured for 16 h with poly (I:C) (Sigma-Aldrich, P0913, 20 µg/mL). The extent of maturation of the DCs was assessed by phenotype analysis. Most of the DCs were mature as they expressed high levels of the DC maturation markers CD83, CD86, and CD80.Citation46 The matured antigen-loaded DCs (mDCs) were washed and co-cultured with T cells.
The iDCs from chronic HBV carrier donors were seeded into 96 well v-bottom plates at 3 × 104 cells/well and cultured for another 24 h (without cytokines). Dialysis buffer, C-HBV, and its components or tetanus toxoid (TT) were added to the cells for 6–8 h. Cells were then incubated with poly (I:C) (20 μg/mL) for 24 h before co-culture with PBMCs.
Isolation of human PBMC-derived T cells
T cells were isolated from PBMCs of uninfected donor by negative selection using Dynal Biotech T cell negative selection kit (Thermo Fisher Scientific, 11344D) as per manufacturer’s procedure. Exceptions were that matched plasma was used instead of BSA and FBS. CD3 + T cells comprised greater than 98% of the isolated population. The T cells were used either labeled with carboxyfluorescein N-succinimidyl ester (CFSE) (see below) or left unlabeled and added directly to cell cultures with antigen-loaded mature DCs.
T cell proliferation assay
Freshly isolated or previously frozen T cells from uninfected donors or PBMCs from chronic HBV carrier donors (in a 15 mL Tube, Corning, C352196) were washed once with D-PBS/5% autologous plasma (AP). The cells were resuspended at 4 × 106 cells/mL in D-PBS/5% autologous plasma (AP) and 110 μL of a 50 μM dilution of CFSE (Thermo Fisher Scientific, 65-0850-84) in D-PBS was added per 1 mL of cells. The cells were mixed and incubated at room temperature for 15 min. After incubation, the tube of cells was topped up to 15 mL with D-PBS/5% AP and the cells were pelleted by centrifugation. The cells were washed twice with 15 mL D-PBS/5% AP. The CFSE-labeled T cells or PBMCs were then resuspended at 4 × 106 cells/mL with AIM V/2.5% AP for co-culture with antigen-loaded mDCs. T cells from the uninfected donor were incubated with antigen-loaded mDCs at ratios of 1–2 × 105 T cells to 2–4 × 104 mDCs per well of a 96 well v-bottom plate in AIM V/2.5% matched serum. T cells were cultured for 7 d. PBMCs from chronic HBV carrier donors were seeded into 96 well plates at 4 × 105 cells/well, incubated for 24 h at 37°C and the PBMCs were stimulated with mDCs loaded with dialysis buffer, C-HBV, its components or TT (tetanus toxoid, EMD Chemicals Inc, 582231). Seven days after stimulation, the CFSE-labeled T cells and PBMCs were washed with D-PBS/2% FBS and stained with fluorescently labeled anti-human-CD3-PE (BD Biosciences, 552127), anti-CD4-APC (BD Biosciences, 555349) and anti-CD8-PerCP (eBioscience, 46-0087-42) antibodies for 20 min on ice. The cells were then washed twice with D-PBS/2% FBS, fixed and acquired using a FACSCalibur flow cytometer. Pentamer binding to activated T cells were analyzed 7 d following the two stimulation APA protocol ().
Detection of intracellular IFN-γ and TNF-α
T cells from the uninfected donor were incubated with antigen loaded mDCs at a ratio of 1–2 × 105 T cells to 2–4 × 104 mDCs for 10 d and then re-stimulated with freshly prepared antigen-loaded mDCs. T cell function was assessed after 6 h of second stimulation. For the determination of intracellular cytokine production, brefeldin A (BD Biosciences, 347688) at 1 µg/mL was added to prevent cytokine release. The production of IFN-γ and TNF-α was quantified using a standard intracellular cytokine labeling protocol (BD Biosciences). In brief, this consisted of labeling the cells with specific fluorochrome-conjugated mAbs for the detection of CD3 (anti-CD3-APC, BD Biosciences, 555335) and CD8 (anti-CD8-PE-Cy5, BD Biosciences, 555368), followed by fixing and permeabilization. Following this, the cell samples were incubated with anti-IFN-γ-PE (BD Biosciences, 562016) and anti-TNF-α-FITC (BD Biosciences, 554512). Between 20,000 and 100,000 cells were acquired per sample using a BD FACSCalibur. The assay protocol is presented in .
PBMCs from chronic HBV-infected donors were seeded into 96 well plates at 4 × 105 cells/well and were co-cultured with antigen-loaded PBMCs for 10 d. PBMCs were co-cultured again (second stimulation) with the mDCs for 30 min at 37°C, at which time 1 μg/mL Golgi Plug (BD Biosciences, 555029) was added, and incubated for an additional 6 h. After 6 h, the cells were washed with D-PBS/2% FBS and stained with antibodies to cell surface markers, anti-CD8 PerCP (BD Biosciences, 347314) and anti-CD3 APC (BD Biosciences, 555335), for 20 min on ice. The cells were washed twice with D-PBS/2% FBS, fixed, permeabilized and stained with anti-TNF-α FITC (BD Biosciences, 554512) and anti-IFN-γ PE (BD Biosciences, 554791) for 30 min on ice. The cells were washed twice with Cytofix/Cytoperm buffer (BD Biosciences, 554714), resuspended in D-PBS/2% FBS/2% PF and acquired using FACSCalibur flow cytometer.
Detection of intracellular GrB and Pfn
The production of GrB and Pfn was quantified using a standard intracellular cytokine labeling protocol (BD Biosciences). In brief, after the second stimulation of T cells from uninfected donor or PBMCs from chronic HBV-infected donors, were labeled with specific fluorochrome-conjugated mAbs for detection CD3 (anti-CD3-APC, BD Biosciences, 555335) and CD8 (anti-CD8-PE-Cy5, BD Biosciences, 555368), followed by fixing and permeabilization. Cells were harvested 4 d after the second stimulation and incubated with anti-GrB-FITC (BD Biosciences, 560211) and anti-Pfn-PE (BD Biosciences, 563763). About 20,000–40,000 cells were acquired per sample using a BD FACSCalibur. The assay protocol is presented in .
Pentamer analysis
Pentamer bindingCitation47 was used to quantify the specificity of CD8 + T cells by using a standard pentamer and intracellular staining protocol (ProImmune and BD Biosciences). T cells from uninfected donor were labeled with anti-CD8-PE-Cy5 (BD Biosciences, 555368), anti-CD3-APC (BD Biosciences, 555335), and the unconjugated HLAA*0201 Pentamer HBV Core FLPSDFFPS (Proimmune, Peptide code 023). The unconjugated Pentamers were detected with a PE-conjugated Fluorotag (ProImmune, K2A-D). Approximately 50,000 cells were acquired using the FACSCalibur. The assay protocol is presented in .
IFN-γ and TNF-α intracellular staining following stimulation with S1, S2, and Core overlapping peptides
PBMCs from chronic HBV carrier donors stimulated for 10 d ex vivo with C-HBV were re-stimulated with overlapping peptides (Mimotopes) of S1, S2 and Core antigens (15 amino acid peptides, 5 amino acid overlap) to induce cytokine production. The peptides were derived from the sequences of PreS1/S2 fragments and core in C-HBV. Peptides were resuspended in 50% acetonitrile and diluted to 1 µM in 0.25% acetonitrile before adding to culture. The PBMCs were incubated for 30 min at 37°C, at which time 1 μg/mL Golgi Plug was added for an additional 6 h of incubation. After 6 h, the cells were washed with D-PBS/2% FBS and stained with antibodies to cell surface markers, anti-CD8-PerCP (BD Biosciences, 321314) and anti-CD3-APC (BD Biosciences, 555335) for 20 min on ice. The cells were washed twice with D-PBS/2% FBS, fixed, permeabilized and stained with anti-TNF-α-FITC (BD Biosciences, 554512) and anti-IFN-γ-PE (BD Biosciences, 554791) for 30 min on ice. The cells were washed twice with perm/wash buffer (BD Biosciences, 554723), resuspended in D-PBS/2% FBS/2% PF and acquired using FACSCalibur flow cytometer.
Analysis of regulatory T cells and apoptosis
The percentage of CD4+ CD25+ regulatory T cells (Treg) or CD4+ CD25-responder T cells (Tresp) producing GrB in PBMCs from chronic HBV-infected donors was assessed by standard phenotype analysis and intracellular staining protocol. For examining the GrB expression in Tresp, the PBMCs were stimulated with C-HBV for 7 d and then labeled with anti-human CD4 – PerCP (BD Biosciences, 347324), anti-human CD25-APC (BD Biosciences, 560987) antibodies. Following cell surface staining, the cells were fixed, permeabilized, and then incubated with anti-human GrB-FITC (BD Biosciences, 560211). Between 30,000 and 40,000 cells were acquired using FACSCalibur flow cytometer. For monitoring apoptosis of Treg cells, the PBMCs were stimulated with C-HBV for 7 d and then labeled with anti-human CD4-PerCP, anti-human CD25-APC antibodies and Annexin V-FITC (BD Biosciences, 556419). The cells were fixed after cell surface staining. Approximately 12,000 cells were acquired using FACSCalibur flow cytometer.
Evaluation of cytolytic activity by DC-based cytotoxic assay using Real-time cell electronic sensing (RT-CES) system
The RT-CES system (xCELLigence, Roche) has been used for assessing a variety of cytotoxicity assays,Citation48–Citation52 including natural killer (NK) cell-mediated cytotoxic activity.Citation53 Briefly, target cells cultured in E-plates are mounted onto a device station placed inside a CO2 incubator. Under the control of RT-CES software, the sensor analyzer automatically monitors cell index (CI), a parameter which represents cell status according to changes in electrode impedance in the wells. Since the CI value is proportional to the culture area covered by attached cells, cell damage and death result in detachment of cells from the wells leading to decrease in CI value. The data of real-time CI were collected and plotted according to experimental analysis. CI at each point is defined as:
where N is the number of the frequency points at which the impedance is measured, and Rb(fi) and Rcell(fi) are the frequency-dependent electrode impedance with or without cells present in the well, respectively. To minimize random error among individual wells, a normalized CI at each point is also calculated by dividing the CI at the time point by the CI at a reference time point. A normalized CI is 1 at the reference time point.
In order to assess C-HBV-induced CTL-mediated cytolytic activity, a DC-based RT-CES cytotoxicity assay was developed. In this assay, antigen-loaded mature DCs derived from human PBMCs of healthy donors or from chronic HBV carriers were used as the target cells. Isolated T cells from uninfected donors or whole PBMCs from chronic HBV-infected donor, activated using the antigen presentation by DCs, were used as effector cells. T cells or PBMCs were co-cultured with C-HBV (or the respective control antigens)-loaded mDCs for 10 d. The target DCs generated from autologous PBMCs were plated into a microelectronic sensor plate (E-plate; 0.6–1 × 105/well) and loaded with protein and its controls followed by maturation with poly (I:C). The E-plate was then put on RT-CES system for monitoring DC growth. On day 10, expanded T cells were harvested from the plate and transferred to corresponding wells in target DCs E-plate (6–8 × 105/well). Changes in CI were dynamically monitored using the RT-CES system. The monitoring was continued for 72 h after the co-culture of T cell/PBMC-target DC. At the end of the 72 h co-culture, the cells were harvested for intracellular staining of GrB production in CD8+ and CD4+ T cells and were evaluated using multi-color flow cytometry.
Results
Production and characterization of C-HBV protein
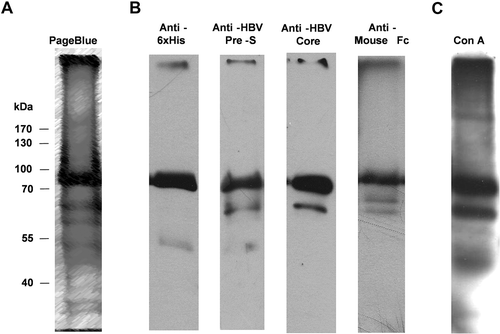
The C-HBV fusion protein containing N-terminal 6xHis tag () was produced in Sf9 insect cells using the baculovirus expression system and purified under denaturing conditions by Ni-chelation affinity chromatography. Following separation by SDS-PAGE under reducing conditions, purified C-HBV appeared as a major band with a molecular weight of ~78 kDa (). Along with the major monomer band, some aggregation products, possibly including the dimer of the protein, was observed. Minor bands of smaller molecular weight proteins, possibly degradation productions, were also seen. Detailed structural studies on the protein have not been carried out. C-HBV protein was characterized by immunoblot using monoclonal antibodies (mAb) recognizing the 6xHis tag, HBV PreS2, HBV Core and murine IgG1 Fc (). Concanavalin A (ConA), a lectin with affinity for terminal α-D-mannosyl residues on glycoproteins, was used to determine if purified C-HBV was glycosylated. Purified protein reacted with ConA () consistent with glycosylation of the C-HBV fusion protein when produced in Sf9 insect cells.
Figure 3. Biochemical characterization of purified C-HBV.
Receptor-mediated binding and internalization of C-HBV by immature DCs
Chimigen® proteins are designed to target Fc receptors on DCs by virtue of the Fc fragment present in the fusion proteins and the glycosylation of the protein would allow binding to C-type lectin receptors on immature DCs (iDCs). To verify the binding of C-HBV to DCs, iDCs were assessed by flow cytometry following incubation with C-HBV (). C-HBV binding to iDCs increased in a concentration-dependent manner. The specific binding to FcγRII and CD206 receptors was determined using blocking mAbs to CD32 and CD206. Compared to buffer control, both anti-CD32 and anti-CD206 inhibited binding by approximately 65% and 46%, respectively (,). Addition of both anti-CD32 and anti-CD206 together, further inhibited C-HBV binding to iDCs ~72% (). The specificity of receptor-binding was further evaluated using confocal microscopy. Anti-CD32 antibody was used to block the binding to CD32 before exposure to C-HBV protein. Blocking the CD32 receptor inhibited binding of the fusion protein to the iDC surface, suggesting that the CD32 receptor is involved in binding of the protein (). Therefore, CD32 and CD206 appear to be major receptors involved in the binding and uptake of C-HBV.
Figure 4. Binding of C-HBV to iDCs-FACS analysis.
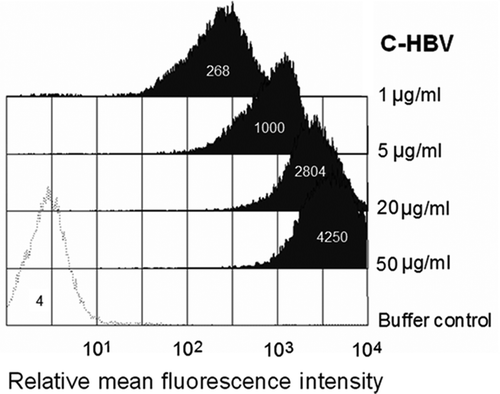
Figure 5. Inhibition of C-HBV binding to iDCs by antibodies to CD32 and CD206.
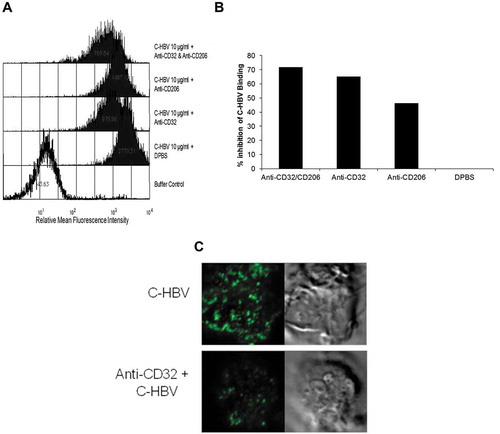
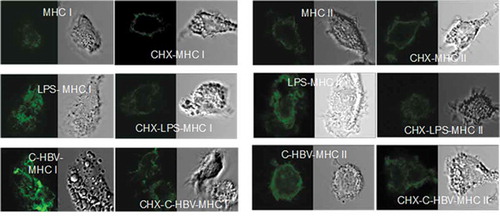
Confocal microscopy also was used to study the binding of C-HBV to iDCs and the role of receptor-mediated internalization, intracellular processing and antigen presentation by DCs. The iDCs incubated with C-HBV at 4°C showed an intense staining on their surface compared with buffer only controls indicating that C-HBV bound to the cell surface (). Internalization of C-HBV by iDCs was evaluated by directly incubating iDCs in media containing C-HBV at 37°C (). Internalization was also studied by initially binding the C-HBV to iDCs at 4°C, removing the unbound protein by washing, and monitoring the internalization of C-HBV by tracking the uptake of the receptor-bound protein at 37°C over time (0, 10, 30, and 60 min). C-HBV localized in punctate staining pattern, often in the vicinity of the cell nuclei, suggesting C-HBV was internalized (,), and binding and internalization increased in a concentration-dependent manner (,). The role of receptor-mediated endocytosis in the uptake of C-HBV was studied by its co-localization with transferrin. Immature DCs were pulsed with C-HBV at 4°C, washed, and incubated for 15 min at 37°C. The cells were fixed, permeabilized and labeled with antibodies to detect C-HBV as well as the transferrin receptor. The transferrin receptor is taken up by receptor-mediated endocytosis and then recycled back to the plasma membrane from early endosomes.Citation54,Citation55 At 4°C, C-HBV bound to the cell surface while the transferrin receptor was present predominantly within cells (). At 37°C, endosomes were observed to form, some of which contained both the C-HBV protein and transferrin receptors. The co-localization of both molecules suggests that C-HBV may be taken up by receptor-mediated endocytosis (). Furthermore, expression of MHC I and MHC II was evaluated by confocal microscopy. Following stimulation with either LPS (positive control) or C-HBV for 24 h, there was a substantial increase in the intensity of MHC I and MHC II on the surface of DCs compared to untreated cells. The co-localization of C-HBV with MHC I and MHC II is shown in . Treatment with the protein synthesis inhibitor cycloheximide inhibited the expression of the MHC I and II which indicated that iDCs synthesized new MHC molecules that migrated to the cell surface after stimulation ().
Figure 6. Binding and internalization of C-HBV by iDCs-confocal microscopy.
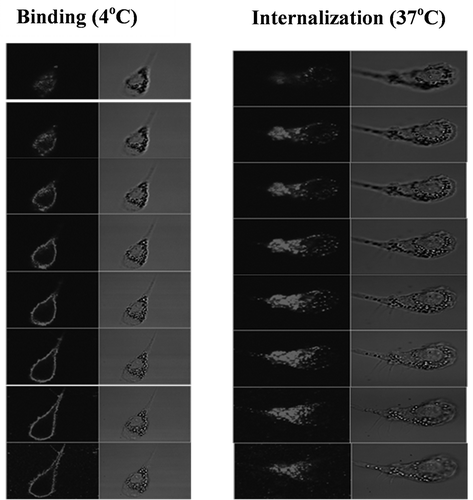
Figure 7. Time and concentration-dependent internalization of C-HBV by iDCs-confocal microscopy.
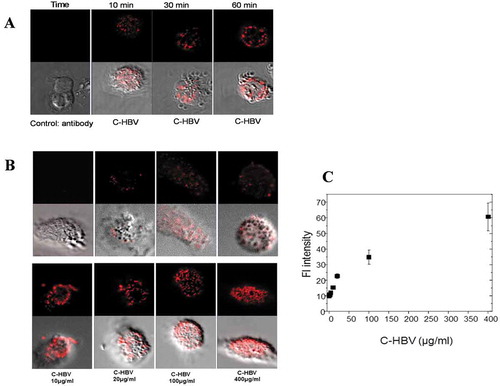
Figure 8. Co-localization of internalized C-HBV and transferrin-confocal microscopy.
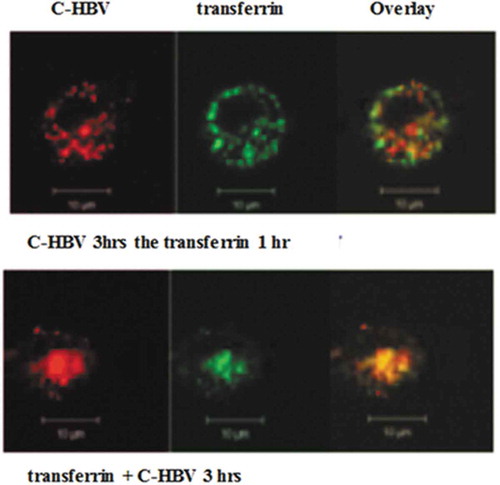
Figure 9. Co-localization of C-HBV and MHC class I & II-confocal microscopy.
T cell responses to C-HBV in healthy uninfected donor cells, ex vivo
The antigen presentation assay (APA) quantifies functional T cell immune responses and the ability of antigen-loaded mature DCs (mDC) to expand antigen-specific T cell clones. Peripheral blood mononuclear cell (PBMC)-derived monocytes from healthy (HBsAg, HBV DNA, and HBcAb-negative) donors were differentiated to iDCs, loaded with antigen and further differentiated into mDCs using Poly (I:C), and these antigen-loaded mDCs were cultured together with autologous naïve T cells. For the analysis of T cell function and specificity, proliferation and production of IFN-γ, TNF-α, perforin (Pfn) and granzyme B (GrB) was assessed following re-stimulation with antigen-loaded mDCs, and peptide-specific responses were assessed using MHC I pentamers.
Both CD4+ and CD8+ CFSE-labeled T cells responded to C-HBV with a higher percentage of proliferation than observed with either dialysis buffer or any of the individual components (HBV S1/S2, HBV core protein or mTBD) (,). Generally, CD4 + T cells displayed a higher percentage of proliferation than CD8 + T cells (,). These results show that antigen presentation of C-HBV by DCs to naïve T cells result in their activation and proliferation.
Figure 10. C-HBV- induced T cell proliferation, production of IFN-γ, TNF-α, GrB and Pfn in T cells.
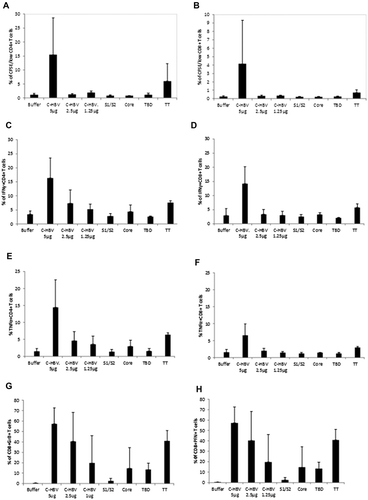
T cell activation was further evaluated by measuring the production of the Th1 cytokines IFN-γ and TNF-α following C-HBV antigen presentation by DCs, as these cytokines were shown to be involved in the elimination of cccDNA without cytolysis.Citation56,Citation57 Compared to buffer and controls (HBV S1/S2, HBV core protein or mTBD), stimulation with C-HBV induced a substantially higher percentage of IFN-γ producing CD4+ and CD8 + T cells (,). Similarly, C-HBV induced a marked increase in the percentage of CD4+ and CD8 + T cells producing TNF-α (,). This further confirmed the ability of C-HBV to activate both CD8+ and CD4 + T cells. The emphasis of the present study was on the cellular response as measured by Th1 cytokines in terms of their relevance to chronic hepatitis B, as originally suggested by Chisari and coworkersCitation58 and recently shown by Xia et al.Citation56 Nevertheless, analysis of Th2 cytokines might have revealed further aspects of the immune response, even if such data do not fit the hypothesis that Th1 response is more relevant to the clearance of chronic hepatitis B. Both Th1 and Th2 responses to C-HBV were evaluated in sheep,Citation59 by measuring lymphocyte proliferation and IFN-γ secretion, as well as the production of HBV antigen-specific antibodies. The results in sheep show a mixed Th1/Th2 response, but nature of the involvement of Th2 cytokines could not be predicted, as this data is not available.
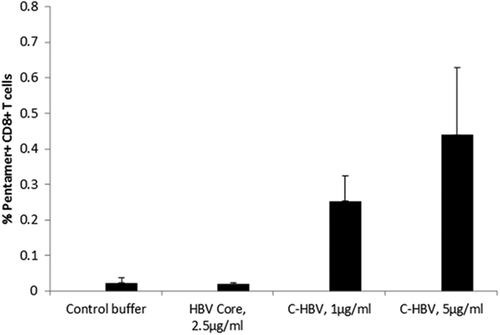
The expression of GrB and Pfn in T cells was also quantified as a means to assess the capability of C-HBV to induce a cytotoxic T cell response. There was marked induction of both GrB and Pfn in CD8 + T cells following stimulation with C-HBV and tetanus toxoid (TT, positive control) compared to HBV S1/S2, HBV core or mTBD (,). Compared to TT and component antigens in the fusion protein, C-HBV induced a higher percentage of CD8 + T cells expressing GrB and Pfn. There was a significant increase in the population of HBV core peptide-specific pentamer-positive CD8 + T cells from C-HBV-stimulated cultures (). The results from the ex vivo APA using cells from healthy HBV un-infected donors show that the C-HBV fusion protein is internalized and processed by DCs promoting proliferation and activation of both CD8+ and CD4 + T cells and differentiation to effector T cells. The data showing internalization of C-HBV, co-localization with MHC I and II, together with our T cell functional data, suggest that the DCs would have processed the protein for presentation in MHC I and II. The identification of core peptide-specific cells by pentamer staining suggests the processing of C-HBV. Furthermore, the recall responses to the peptide pools indirectly suggest that the processed peptides from HBV antigens were instrumental in defining the specificity of the activated T cells.
Figure 11. Generation of HBV core peptide pentamer-specific T cells.
T cell responses to C-HBV in PBMCs isolated from chronic HBV carriers
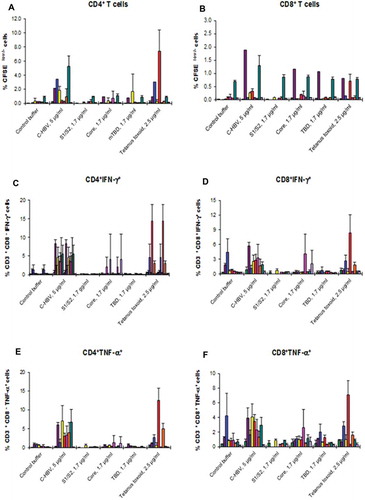
Having shown the nature of the action of C-HBV in cells from HBV un-infected donors, the effect of C-HBV in PBMCs derived from chronically HBV-infected individuals was tested. Chronic HBV infection results in T cell exhaustion.Citation12 An effective immunotherapy for chronic HBV would ideally restore HBV-specific T cell responses. The ability of C-HBV to induce T cell responses was evaluated using the APA with PBMCs-derived DCs and T cells from chronic HBV carriers. T cells isolated from chronically HBV-infected individual donors (n = 9) stimulated ex vivo with C-HBV-pulsed DCs induced proliferation of both CD4+ and CD8+ CFSE-labeled T cells compared to the respective controls (dialysis buffer, HBV S1/S2, HBV core protein or mTBD) (,). With cells isolated from healthy HBV-seronegative donors, CD4 + T cells displayed a higher percentage of proliferation than CD8 + T cells (,). Similarly, using the APA ex vivo, after two stimulations of PBMCs, C-HBV induced a higher percentage of IFN-γ+ (,) and TNF-α+ (,) producing CD4+ and CD8 + T cells than either the buffer control, or any of the components of C-HBV (HBV S1/S2, HBV core protein or mTBD). The secretion of IFN-γ by PBMCs from chronically HBV-infected donors also showed similar pattern (data not presented). These results demonstrate that C-HBV is able to induce T cell Th1 cytokine production in PBMCs from chronically HBV-infected individuals.
Figure 12. Induction of T cell proliferation and production of IFN-γ and TNF-α positive T cells by C-HBV in PBMCs from chronically HBV-infected donors.
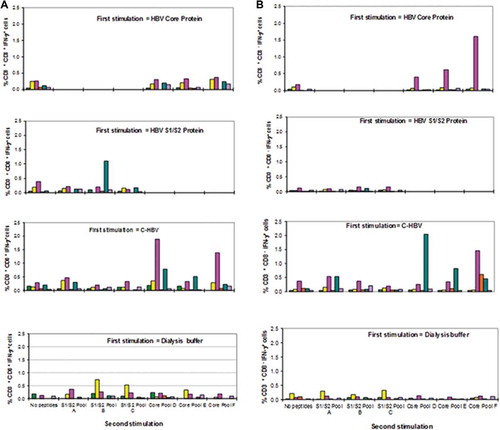
C-HBV peptide epitope-specificity of T cell responses
The specificity of the CD8+/CD4 + T cells generated in the ex vivo assays were assessed following the re-stimulation of C-HBV-primed PBMCs with overlapping peptide pools containing HBV S1/S2 fragments or core sequences. The pools containing peptides (15 amino acid, 5 amino acid overlap, 9–14 peptides per pool), were used for each of the HBV antigens (S1/S2/Core). PBMCs from chronic HBV carriers (n = 6) were cultured in the presence of C-HBV, S1/S2, core antigen or buffer and recall responses were then assessed following incubation of cells with the overlapping peptide pools from HBV S1/S2 fragments and core antigen by determining the percentage of IFN-γ producing CD8+ and CD4 + T cells. The PBMCs which were treated with C-HBV showed recall responses to many of the peptide pools (,). In general, responses were more pronounced with peptides derived from HBV core, although S1/S2 peptides showed a recall response. These results suggest that C-HBV is able to induce HBV-specific T cell response, directed against multiple epitopes, especially within HBV core, shown to be important for the resolution of chronic HBV infection.Citation3
Figure 13. HBV peptide-specific T cell recall responses in chronic HBV carrier PBMC-derived T cells activated by C-HBV.
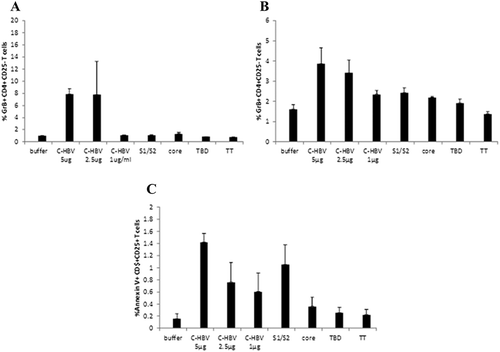
Induction of functional responder T cells (T resp) by C-HBV
Regulatory T cells (Treg) have been shown to contribute to T cell dysfunction and HBV persistence by suppressing virus-specific T cell responses in chronically HBV-infected patients.Citation15 Activated CD4 responder T cells (Tresp, CD4+ CD25-) were shown to produce GrB and actively remove effector regulatory T cells (Treg, CD4+ CD25+ FoxP3+) by apoptosis.Citation60 PBMCs isolated from two representative donors (, donor #14) and (, donor #23) were stimulated ex vivo with C-HBV and the respective controls. There was a significant increase in the GrB production seen in the C-HBV treated group (,). These results demonstrate that C-HBV is able to induce GrB production in Tresp cells in PBMCs from chronically HBV-infected individuals. Staining for Annexin V+ cells was used to monitor the apoptosis of Treg cells by activated Tresp cells. There was an increase in Annexin V stained cells in CD4+ CD25+ cells (Treg phenotype) from C-HBV-stimulated PBMCs compared to the respective controls in chronic carrier PBMCs (). These results suggest that the apoptosis of Tregs may be mediated by C-HBV-activated Tresp cells and may possibly play a role in breaking tolerance to chronic HBV infection.
Figure 14. Production of GrB positive T responder cells by C-HBV in PBMCs from chronic HBV carriers. PBMCs from donor CHBV#14 (Panel A) and #23 (Panel B) were stimulated ex vivo with either dialysis buffer, C-HBV or components for 7 d. The percentage GrB+ CD4+ CD25-T responder cells (Tresp) was determined by intracellular cytokine staining and flow cytometry. A batch of PBMCs from donor #23 was stained for Annexin V. The percentage of CD4+ CD25+ Annexin V+ (Treg cell phenotype) was determined by flow cytometry (Panel C). Each bar represents results from average of triplictae wells from the same group.
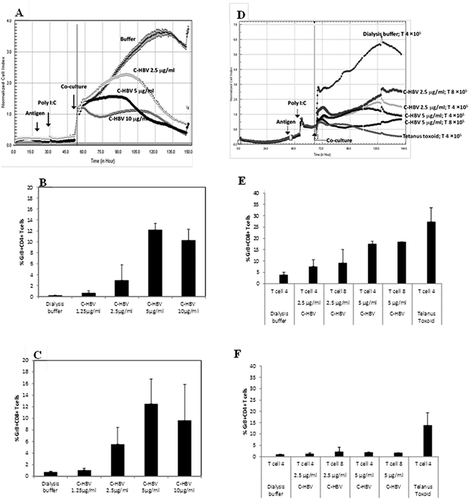
CTL-mediated cytolytic activity induced by C-HBV
Cell lysis under various conditions,Citation48–Citation52,Citation61 including natural killer (NK) cell-mediated cytolytic activityCitation53 can be assessed using a real-time cell electronic sensing (RT-CES) system (xCELLigence, Roche). A DC-based RT-CES cytolytic assay was developed to study the effects of C-HBV on the induction of T cell-mediated cell killing using cells derived from PBMCs isolated from an HBV un-infected healthy donor (control) and HBV chronic carriers (n = 6). In this assay, PBMC-derived mDCs, loaded with different concentrations of C-HBV or buffer, were used as target cells, and T cells isolated from an uninfected donor or whole PBMCs from chronic HBV carriers were used as effector cells. Lysis of the target cell monolayer was monitored as described in the methods section. Cell indices from an un-infected donor () and a representative HBV-carrier are presented (). The cell killing was similar in the other HBV-carrier samples (data not shown). The expression of GrB among the T cell population in un-infected (,) and in HBV carrier (,) is shown. The decrease in cellular index as a function of time, both in the un-infected and the HBV-infected donor cells, and the expression of GrB, suggests that C-HBV is able to promote the induction of cytotoxic T cell responses.
Figure 15. RT-CES dynamic monitoring of apoptosis by C-HBV-activated T cells.
Discussion
Chronic hepatitis B remains a major healthcare concern worldwide, and in spite of the availability of very effective prophylactic vaccines over 250 million people are estimated to remain chronically infected.Citation1 Currently available antiviral agents can efficiently control virus replication but the induction of durable off-treatment responses remains unattainable in the vast majority of chronic hepatitis B carriers. Many novel therapies that target the HBV life cycle and host immune response are currently in various stages of clinical development.Citation19,Citation62 Strategies to develop agents for functional cure of hepatitis B include the use of novel adjuvants and routes of administration,Citation63 HBV antigens produced in situ using vector plasmids,Citation64,Citation65 various RNAi technologies,Citation66 Toll-like receptor (TLR) effectors,Citation67 mAbsCitation68 and inhibitors of core capsid assembly.Citation69
The mechanisms HBV uses to avoid immune surveillance are not well understood.Citation70 A high viral load and the resulting increase in the concentration of viral antigens may make the immune system tolerant to the virus. Predictors of responsiveness to interferon therapy include a low baseline viral load, high ALT levels, younger age and HBV genotype amongst others.Citation71 In pivotal clinical trials of de novo combination therapy of pegylated interferon with lamivudine,Citation72–Citation74 adefovir,Citation75 or entecavirCitation76 did not improve sustained virological response rates compared to monotherapy but recently interferon combined with tenofovir has been shown to improve post-treatment HBsAg clearance rates during longer-term follow-up,Citation77,Citation78 and in long-term nucleoside/nucleotide suppressed patients a switch to interferon or interferon “add on” therapy may enhance response rates in selected patients.Citation71,Citation79 This suggests that interferon-mediated immune clearance may be more effective in clearing the virus when the viral load has been reduced for extended periods.
Many mechanisms may contribute to the development of HBV tolerance including an immunosuppressive role mediated by regulatory T cells (Tregs) which exert suppressive function on immune responses that may cause persistent HBV infectionCitation80 and the suppressive function of Tregs can be enhanced by virus infection.Citation81 A number of studies have shown that Tregs in PBMCs isolated from HBV-infected patients inhibited HBV-specific immune response.Citation82–Citation84 These results suggest that the modulation of Treg might be a potential therapeutic strategy for the treatment of chronically infected patients. The lack of proper presentation of the appropriate viral antigen to the host immune system may also be a contributing factor. It has been reported that the functional defects of antigen-presenting cells, such as DCs, may cause the impaired T cell response during HBV infection.Citation22,Citation85
Although treatment regimens for chronic hepatitis B have markedly improved outcomes during the last decade, the need for immune system activation to effectively induce sustained off-treatment disease remission and clearance of the hepatitis B surface antigen (‘functional cure’) still remains a largely unachieved target.Citation86,Citation87 The host immune system plays a dominant role in the elimination of HBV infection. In acute HBV infections in adults, more than 90% of the infections can be cleared by the host immune system, whereas in infants with the immune system not fully functional the clearance rate is less than 10%.Citation11,Citation88,Citation89 During clinical trials of lamivudine, it was noted that patients with elevated liver enzymes, a measure of immune system stimulation, cleared the HBV infection at a rate greater than patients with normal liver enzymes.Citation90 This observation emphasizes the need for the host immune system to be activated against viral antigens which are present in the chronic carrier which the immune system regards as “self” rather than “foreign”. Since impaired immune responses (immune tolerance/ignorance) prevail in chronic HBV carriers, any immunotherapy approach must be able to re-educate the host immune system to recognize the virus and the viral antigens as “foreign” (to break tolerance/ignorance) so that newly activated CTL can clear infected cells and the humoral response can eliminate the circulating antigens.Citation17
The correlation between immune responses and the spontaneous clearance of chronic HBV infection supports the renewed interest in developing HBV immunotherapy agents.Citation21 These include antibodies, protein, and peptides with adjuvants, DNA and viral-vectored molecules which have been used in combination with currently available antiviral agents to decrease viremia. So far, none of these approaches have produced the expected results, and various explanations have been provided to explain the lack of efficacy of these molecules.Citation21,Citation91
A number of therapeutic product candidates have been listed in a recent publication by Boni et al.Citation21 The most advanced product candidate ABX 203 had advanced to Phase III clinical trial. ABX 203 is a mixture of 2 Virus Like Particles (VLPs) from HBsAg and HBcAg. The HBsAg is purified from Pichia pastoris, whereas the HBcAg is purified from E. coli. The vaccine is nasally and subcutaneously administered.Citation63 This trial was recently terminated based on a futility analysis. Heat-inactivated whole yeast particles (Tarmogens) expressing HBV X, surface and core antigensCitation92 was used in a Phase II clinical trial. As there was no significant decrease in serum HbsAg, the development was terminated due to lack of clinical efficacy.Citation93 One of the observations was that, although there was CD8 + T cell activation, there was neither CD4 + T cell activation nor antibody production. It is possible that the immune reaction to the large number of yeast particles overwhelmed the host immune system, with minimal response to the expressed HBV proteins. Yeast Immune Complex (YIC), an “Antigen-antibody (HBsAg-HBIG) immunogenic complex” with Alum as adjuvant, did not show clinical efficacy in a Phase III clinical trial.Citation94 The observation that “placebo” group showed superior response than treated group, implied lack of efficacy. Among the vectored vaccines, TG1050 has completed Phase Ib trial.Citation95 TG1050 is an adenovirus serotype 5 vectored vaccine encoding truncated HBV core, a modified HBV DNA polymerase and two HBV envelope domains. IFN-γ producing HBV-specific T cells were produced, but only minor decreases in HBsAg was observed. Further clinical trials using a larger number of patients would provide a better understanding of the efficacy. The major issue in using adenoviral vector is the preexisting immunity against the vector, which can abrogate any clinical efficacy.Citation96–Citation98
It has been reported that adjuvant-based vaccines can induce specific T cell responses to cancer cells, but most of the antigen-specific T-cells were sequestered, dysfunctional and deleted at the vaccination site due to the persisting “depot effect” of the adjuvanted vaccineCitation99 which is a possible explanation for the lack of efficacy of some of the listed HBV therapeutic candidates.
The success in restoring effective antigen-specific immune control of the viral infection may depend on various strategies including those that target antigen processing and presentation by the APCs for effective generation of CTLs, down-regulation of Tregs and induction of effective humoral responses.
The technology described herein employs the use of Fc fusion proteins to target DC receptors for antigen delivery and activation of immune responses. This platform technology combines the benefits of both recombinant (neo)-antigens and the xenotypic antibody in inducing a broad and antigen-specific immune response. Targeted delivery of antigens to DCs has been a challenge, although several studies have shown that antigen-targeting to DCs can be employed to enhance T cell priming and induction of effector T cell and humoral responses.Citation28,Citation100 The fusion protein (C-HBV) is produced in Sf9 insect cells which have less complex N-glycosylation (terminal mannose, no sialic acid) than eukaryotic cells,Citation32 switching the nature of the protein while maintaining the primary amino acid sequence intact. The glycosylation targets lectin receptors on DCs and the primitive nature of the glycosylation makes the whole molecule highly immunogenic in comparison to the mammalian or yeast-derived HBV antigens. Chimigen® Technology is designed such that the protein specifically targets DC receptors so that it is properly processed and presented by MHC I and II to generate multi-antigen, multi-epitope-specific T and B cell immune responses. Chimigen® Technology is a unique approach to vaccine and immunotherapy development which is expected to avoid many of the drawbacks seen with previous attempts.
Various antibody-based therapies partially achieve this goal by binding to the respective antigen and the antigen-antibody complex entering the DCs via receptor-mediated uptake.Citation101 For example, Trastuzumab (Herceptin), a humanized IgG1 k mAb that binds to the extracellular domain of epidermal growth factor 2 (HER2), apart from inhibition of HER2 downstream signaling and antibody-dependent cell-mediated cytotoxicity, has recently been shown to enhance FcR-mediated uptake and cross presentation by HER2-derived peptides by DCs, thereby enhancing the generation of peptide-specific CTL.Citation102 There are a large number of clinical trials currently in progress using mAb for various cancer and infectious diseases applications.Citation103–Citation105 Antibody-based therapies require the antigen to be complexed in situ for efficient delivery to DCs. With levels of circulating antigens varying among individuals the efficiency of uptake by the DCs also depends on the nature of the complex. Aggregates of the complex could be engulfed by scavenger cells and macrophages resulting in a lack of appropriate antigen presentation.Citation106 Antigen-antibody complexes can bind to DC receptors which result in appropriate antigen uptake, processing and productive presentation, whereas aggregates may be taken up via micropinocytosis for degradation in lysosomes resulting in poor antigen presentation.Citation106 Another problem is that if the antibody is in excess of antigen, T cell responses can be inhibited.Citation107 In antibody-based therapies, the administered antibody concentration has to be adjusted to each patient to optimally complex antibody with circulating autologous antigen.Citation108 In the case of Chimigen® Technology, this variable is avoided since the stoichiometry of antigen/antibody is maintained in the protein molecule. An added advantage is that this does not have to rely on circulating antigen for presentation.
C-HBV is a protein immunotherapy with no adjuvant and therefore, the depot effect is avoided.Citation99 Avoiding a depot effect of adjuvant would thus be a definite advantage. Unlike a protein administered with adjuvant, C-HBV would not be expected to remain at the site of injection but instead, be readily taken up by DCs as observed in a large animal model (sheep) following C-HBV injection.Citation59
The generation of HBV-specific T cells (IFN-γ, TNF-α, GrB, Pfn +ve) is the most relevant for the predicted anti-HBV immunotherapeutic effect. Further, the ex vivo assays demonstrated that not only do T cells proliferate, but they also differentiate into effector cells secreting antiviral cytokines and mediators of cytolytic activity.
C-HBV contained N-terminal 6xHis tag to facilitate the purification using Ni-chelate affinity chromatography. The protein showed no toxic effect either in ex vivo studies using human PBMCs, or in vivo studies using sheep.Citation59 Furthermore, the antibody responses observed against the 6xHis were of much lower magnitude than any of the HBV antigens. The presence of the tag could be an issue if used in clinical trials but there are products with His tag that have reached Phase 2, including one for HBV,Citation92,Citation93 with no safety issue identified and none has reached Phase 3 so far.
One of the limitations of the current study is the lack of Th2 cytokine data. The present study focused on the induction of Th1 immune responses by C-HBV, measured as the production of Th1 cytokines IFN-γ and TNF-α. The rationale was to assess for markers of cellular immunity and these were the most relevant.Citation56,Citation58 Analysis of Th2 cytokines might have provided additional information on the immune responses, even if such data would not fit the current hypothesis that Th1 responses are more meaningful in chronic hepatitis B treatment. Although the present study shows compelling results ex vivo in human cells for an effective immunotherapy for chronic hepatitis B, one other limitation of the study is that the clinical efficacy of this approach remains to be proven. The next step is to test C-HBV in clinical trials in humans.
Conclusion
The experimental results presented in this report demonstrate that C-HBV binds to specific receptors CD32 and CD206 on DCs, and subsequently is internalized and processed for presentation on both MHC I and II. Presentation of C-HBV to T cells from HBV un-infected healthy donors or chronic HBV carriers resulted in the proliferation of CD4+ and CD8 + T cells and the production of IFN-γ, TNF-α, GrB and Pfn, consistent with CTL function. The data presented herein provides “proof of concept” of the Chimigen® Technology platform for DC-receptor targeted delivery of antigen(s) and subsequent induction of effective CD4+ and CD8 + T cell responses. The induction of CD4 + T cells from chronic HBV carrier PBMCs along with the production of T responder cells and the targeted removal of cells with a T regulatory phenotype provides preliminary supportive evidence for a role of C-HBV to break immune tolerance to HBV.
Abbreviations
| APA | = | Antigen presentation assay |
| AF | = | Alexa Fluor |
| APC | = | Allophycocyanin |
| C-HBV | = | Chimigen® HBV |
| CFSE | = | Carboxyfluorescein N-succinimidyl ester |
| Cy | = | Cyanine |
| DC | = | Dendritic cells |
| FACS | = | Fluorescence activated cell sorting |
| FITC | = | Fluorescein isothiocyanate |
| HBV | = | Hepatitis B virus |
| IFN-γ | = | Interferon gamma |
| GrB | = | Granzyme B |
| Pfn | = | Perforin |
| PE | = | Phycoerythrin |
| PBMC | = | Peripheral blood mononuclear cell |
| PerCP | = | Peridinin-chlorophyll-protein |
| TNF-α | = | Tumor necrosis factor alpha |
Disclosure of potential conflict of interest
The authors, RG, AM, BM, YES and KG declare they have no conflict of interest.
Acknowledgments
The authors thank Drs. Nils O. Petersen and Chunhong Tian, National Institute for Nanotechnology (NINT), University of Alberta for the confocal microscopy. The assistance from Drs. D. Wang and Y. Xia is gratefully acknowledged. The authors thank Roche Applied Science for the use of xCELLigence RT-CES equipment. The authors thank Rohit George for his assistance with the preparation of the manuscript.
Additional information
Funding
Related Research Data
References
- World Health Organization. Global hepatitis report; 2017. www.who.int/hepatitis/publications/global-hepatitis-report2017/en/.
- Gerlich WH. Prophylactic vaccination against hepatitis B: achievements, challenges and perspectives. Med Microbiol Immunol. 2015;204:39–55.
- Lau GK, Suri D, Liang R, Rigopoulou EI, Thomas MG, Mullerova I, Nanji A, Yuen ST, Williams R and Naoumov NV. Resolution of chronic hepatitis B and anti-HBs seroconversion in humans by adoptive transfer of immunity to hepatitis B core antigen. Gastroenterology. 2002;122(3):614–24. doi:10.1053/gast.2002.31887.
- Rehermann B, Lau D, Hoofnagle JH and Chisari FV. Cytotoxic T lymphocyte responsiveness after resolution of chronic hepatitis B virus infection. J Clin Invest. 1996;97(7):1655–65. doi:10.1172/JCI118592.
- Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH and Chisari FV. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol. 2003;77(1):68–76. doi:10.1128/JVI.77.1.68-76.2003.
- Gish RG, Given BD, Lai CL, Locarnini SA, Lau JY, Lewis DL and Schluep T. Chronic hepatitis B: virology, natural history, current management and a glimpse at future opportunities. Antiviral Res. 2015;121:47–58. doi:10.1016/j.antiviral.2015.06.008.
- Sun D, Zhu L, Yao D, Chen L, Fu L and Ouyang L. Recent progress in potential anti-hepatitis B virus agents: structural and pharmacological perspectives. Eur J Med Chem. 2018;147:205–17. doi:10.1016/j.ejmech.2018.02.001.
- Asselah T, Marcellin P and Schinazi RF. Treatment of hepatitis C virus infection with direct-acting antiviral agents: 100% cure? Liver Int. 2018;38(Suppl 1):7–13. doi:10.1111/liv.13673.
- Guo JT, Guo H. Metabolism and function of hepatitis B virus cccDNA: implications for the development of cccDNA-targeting antiviral therapeutics. Antiviral Res. 2015;122:91–100. doi:10.1016/j.antiviral.2015.08.005.
- Rehermann B. Chronic infections with hepatotropic viruses: mechanisms of impairment of cellular immune responses. Semin Liver Dis. 2007;27(2):152–60. doi:10.1055/s-2007-979468.
- Rehermann B, Thimme R. Insights from antiviral therapy into immune responses to hepatitis B and C virus infection. Gastroenterology. 2019;156(2):369–83. doi:10.1053/j.gastro.2018.08.061.
- Mueller SN, Ahmed R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2009;106(21):8623–28. doi:10.1073/pnas.0809818106.
- Cho H, Kang H, HH L, CW K. Programmed cell death 1 (PD-1) and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) in viral hepatitis. Int J Mol Sci. 2017;18(7):1517. doi:10.3390/ijms18071517
- Fisicaro P, Barili V, Montanini B, Acerbi G, Ferracin M, Guerrieri F, Salerno D, Boni C, Massari M, Cavallo MC, et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat Med. 2017;23(3):327–36.
- Jung MK, Shin EC. Regulatory T cells in hepatitis B and C virus infections. Immune Netw. 2016;16(6):330–36. doi:10.4110/in.2016.16.6.330.
- Wang FS, Xing LH, Liu MX, Zhu CL, Liu HG, Wang HF and Lei ZY. Dysfunction of peripheral blood dendritic cells from patients with chronic hepatitis B virus infection. World J Gastroenterol. 2001;7(4):537–41. doi:10.3748/wjg.v7.i4.537.
- Gehring AJ, Protzer U. Targeting innate and adaptive immune responses to cure chronic HBV infection. Gastroenterology. 2019;156(2):325–37. doi:10.1053/j.gastro.2018.10.032.
- Bertoletti A, Le Bert N. Immunotherapy for chronic hepatitis B virus infection. Gut Liver. 2018;12:497–507.
- Block TM, Rawat S and Brosgart CL. Chronic hepatitis B: a wave of new therapies on the horizon. Antiviral Res. 2015;121:69–81. doi:10.1016/j.antiviral.2015.06.014.
- Dawood A, Abdul Basit S, Jayaraj M and Gish RG. Drugs in development for hepatitis B. Drugs. 2017;77(12):1263–80. doi:10.1007/s40265-017-0769-2.
- Boni C, Barili V, Acerbi G, Rossi M, Vecchi A, Laccabue D, Penna A, Missale G, Ferrari C, Fisicaro P. HBV immune-therapy: from molecular mechanisms to clinical applications. Int J Mol Sci. 2019;20:11. doi:10.3390/ijms20112754.
- Kosinska AD, Bauer T and Protzer U. Therapeutic vaccination for chronic hepatitis B. Curr Opin Virol. 2017;23:75–81. doi:10.1016/j.coviro.2017.03.011.
- Bertoletti A, Gehring AJ. Immune therapeutic strategies in chronic hepatitis B virus infection: virus or inflammation control? PLoS Pathog. 2013;9(12):e1003784. doi:10.1371/journal.ppat.1003784.
- Liang TJ, Block TM, McMahon BJ, Ghany MG, Urban S, Guo JT, Locarnini S, Zoulim F, Chang KM and Lok AS. Present and future therapies of hepatitis B: from discovery to cure. Hepatology. 2015;62(6):1893–908. doi:10.1002/hep.28025.
- Steinman RM, Inaba K, Turley S, Pierre P and Mellman I. Antigen capture, processing, and presentation by dendritic cells: recent cell biological studies. Hum Immunol. 1999;60(7):562–67. doi:10.1016/S0198-8859(99)00030-0.
- Martinez-Pomares L. The mannose receptor. J Leukoc Biol. 2012;92(6):1177–86. doi:10.1189/jlb.0512231.
- Guilliams M, Bruhns P, Saeys Y, Hammad H and Lambrecht BN. The function of Fcgamma receptors in dendritic cells and macrophages. Nat Rev Immunol. 2014;14(2):94–108. doi:10.1038/nri3582.
- Caminschi I, Lahoud MH and Shortman K. Enhancing immune responses by targeting antigen to DC. Eur J Immunol. 2009;39(4):931–38. doi:10.1002/eji.200839035.
- Eickhoff S, Brewitz A, Gerner MY, Klauschen F, Komander K, Hemmi H, Garbi N, Kaisho T, Germain RN and Kastenmuller W. Robust anti-viral immunity requires multiple distinct T cell-dendritic cell interactions. Cell. 2015;162(6):1322–37. doi:10.1016/j.cell.2015.08.004.
- Kastenmuller W, Kastenmuller K, Kurts C and Seder RA. Dendritic cell-targeted vaccines–hope or hype? Nat Rev Immunol. 2014;14(10):705–11. doi:10.1038/nri3727.
- Altmann F, Staudacher E, Wilson IB and Marz L. Insect cells as hosts for the expression of recombinant glycoproteins. Glycoconj J. 1999;16(2):109–23. doi:10.1023/A:1026488408951.
- Shi X, Jarvis DL. Protein N-glycosylation in the baculovirus-insect cell system. Curr Drug Targets. 2007;8(10):1116–25. doi:10.2174/138945007782151360.
- Urbanowicz RA, Wang R, Schiel JE, Keck ZY, Kerzic MC, Lau P, Rangarajan S, Garagusi KJ, Tan L, Guest JD, et al. Antigenicity and immunogenicity of differentially glycosylated hepatitis C virus E2 envelope proteins expressed in mammalian and insect cells. J Virol. 2019;93:7. doi:10.1128/JVI.01403-18.
- Al-Barwani F, Young SL, Baird MA, Larsen DS and Ward VK. Mannosylation of virus-like particles enhances internalization by antigen presenting cells. PLoS One. 2014;9(8):e104523. doi:10.1371/journal.pone.0104523.
- Lohr HF, Krug S, Herr W, Weyer S, Schlaak J, Wolfel T, Gerken G, Meyer Zum Buschenfelde KH. Quantitative and functional analysis of core-specific T-helper cell and CTL activities in acute and chronic hepatitis B. Liver. 1998;18(6):405–13. doi:10.1111/j.1600-0676.1998.tb00825.x.
- Nystrom J, Chen A, Frelin L, Ahlen G, Koh S, Brass A, Peterson DL, Fons M, Milich DR, Hultgren C, et al. Improving on the ability of endogenous hepatitis B core antigen to prime cytotoxic T lymphocytes. J Infect Dis. 2010;201(12):1867–79. doi:10.1086/651178.
- Bertoletti A, Chisari FV, Penna A, Guilhot S, Galati L, Missale G, Fowler P, Schlicht HJ, Vitiello A, Chesnut RC, et al. Definition of a minimal optimal cytotoxic T-cell epitope within the hepatitis B virus nucleocapsid protein. J Virol. 1993;67(4):2376–80.
- Chen X, Li M, Le X, Ma W and Zhou B. Recombinant hepatitis B core antigen carrying preS1 epitopes induce immune response against chronic HBV infection. Vaccine. 2004;22(3–4):439–46. doi:10.1016/j.vaccine.2003.07.014.
- Gruener NH, Gerlach TJ, Ulsenheimer A, Diepolder HM, Wierenga E, Zachoval R, Heeg M, Pape GR and Jung MC. Characterization of sequence variations in immunodominant regions of the HBV-nucleocapsid protein as a prerequisite for the development of an epitope-based vaccine. Vaccine. 2007;25(26):4960–66. doi:10.1016/j.vaccine.2006.12.009.
- Milich DR, McLachlan A, Moriarty A and Thornton GB. Immune response to hepatitis B virus core antigen (HBcAg): localization of T cell recognition sites within HBcAg/HBeAg. J Immunol. 1987;139:1223–31.
- Yang BF, Zhao HL, Xue C, Xiong XH, Zhang W, Yao XQ and Liu ZM. Recombinant heat shock protein 65 carrying hepatitis B core antigen induces HBcAg-specific CTL response. Vaccine. 2007;25(22):4478–86. doi:10.1016/j.vaccine.2007.03.020.
- Cai X, Zheng W, Pan S, Zhang S, Xie Y, Guo H, Wang G, Li Z and Luo M. A virus-like particle of the hepatitis B virus preS antigen elicits robust neutralizing antibodies and T cell responses in mice. Antiviral Res. 2018;149:48–57. doi:10.1016/j.antiviral.2017.11.007.
- Milich DR, McLachlan A, Chisari FV, Kent SB and Thorton GB. Immune response to the pre-S(1) region of the hepatitis B surface antigen (HBsAg): a pre-S(1)-specific T cell response can bypass nonresponsiveness to the pre-S(2) and S regions of HBsAg. J Immunol. 1986;137:315–22.
- Whiteside TL, Stanson J, Shurin MR and Ferrone S. Antigen-processing machinery in human dendritic cells: up-regulation by maturation and down-regulation by tumor cells. J Immunol. 2004;173(3):1526–34. doi:10.4049/jimmunol.173.3.1526.
- Pickl WF, Majdic O, Kohl P, Stockl J, Riedl E, Scheinecker C, Bello-Fernandez C and Knapp W. Molecular and functional characteristics of dendritic cells generated from highly purified CD14+ peripheral blood monocytes. J Immunol. 1996;157:3850–59.
- Zhou LJ, Tedder TF. CD14+ blood monocytes can differentiate into functionally mature CD83+ dendritic cells. Proc Natl Acad Sci U S A. 1996;93(6):2588–92. doi:10.1073/pnas.93.6.2588.
- Davis MM, Altman JD and Newell EW. Interrogating the repertoire: broadening the scope of peptide-MHC multimer analysis. Nat Rev Immunol. 2011;11(8):551–58. doi:10.1038/nri3020.
- Atienza JM, Yu N, Kirstein SL, Xi B, Wang X, Xu X and Abassi YA. Dynamic and label-free cell-based assays using the real-time cell electronic sensing system. Assay Drug Dev Technol. 2006;4(5):597–607. doi:10.1089/adt.2006.4.597.
- Xing JZ, Zhu L, Gabos S and Xie L. Microelectronic cell sensor assay for detection of cytotoxicity and prediction of acute toxicity. Toxicol In Vitro. 2006;20(6):995–1004. doi:10.1016/j.tiv.2005.12.008.
- Xing JZ, Zhu L, Jackson JA, Gabos S, Sun XJ, Wang XB and Xu X. Dynamic monitoring of cytotoxicity on microelectronic sensors. Chem Res Toxicol. 2005;18(2):154–61. doi:10.1021/tx049721 s.
- Boyd JM, Huang L, Xie L, Moe B, Gabos S and Li XF. A cell-microelectronic sensing technique for profiling cytotoxicity of chemicals. Anal Chim Acta. 2008;615(1):80–87. doi:10.1016/j.aca.2008.03.047.
- Xia Y, Xing JZ and Krukoff TL. Neuroprotective effects of R,R-tetrahydrochrysene against glutamate-induced cell death through anti-excitotoxic and antioxidant actions involving estrogen receptor-dependent and -independent pathways. Neuroscience. 2009;162(2):292–306. doi:10.1016/j.neuroscience.2009.04.068.
- Zhu J, Wang X, Xu X and Abassi YA. Dynamic and label-free monitoring of natural killer cell cytotoxic activity using electronic cell sensor arrays. J Immunol Methods. 2006;309(1–2):25–33. doi:10.1016/j.jim.2005.10.018.
- Embgenbroich M, Burgdorf S. Current concepts of antigen cross-presentation. Front Immunol. 2018;9:1643. doi:10.3389/fimmu.2018.01643.
- Guagliardi LE, Koppelman B, Blum JS, Marks MS, Cresswell P and Brodsky FM. Co-localization of molecules involved in antigen processing and presentation in an early endocytic compartment. Nature. 1990;343(6254):133–39. doi:10.1038/343133a0.
- Xia Y, Stadler D, Lucifora J, Reisinger F, Webb D, Hosel M, Michler T, Wisskirchen K, Cheng X, Zhang K, et al. Interferon-gamma and tumor necrosis factor-alpha produced by T cells reduce the HBV persistence form, cccDNA, without cytolysis. Gastroenterology. 2016;150(1):194–205. doi:10.1053/j.gastro.2015.09.026.
- Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R and Chisari FV. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284(5415):825–29. doi:10.1126/science.284.5415.825.
- Yang PL, Althage A, Chung J, Maier H, Wieland S, Isogawa M and Chisari FV. Immune effectors required for hepatitis B virus clearance. Proc Natl Acad Sci U S A. 2010;107(2):798–802. doi:10.1073/pnas.0913498107.
- George R, Ma A, Motyka B, Shi YE, Liu Q, Griebel P. A dendritic cell-targeted chimeric hepatitis B virus immunotherapeutic vaccine induces both cellular and humoral immune responses in vivo manuscript submitted to human vaccines and immunotherapeutics. Hum Vaccin Immunother. 2019 Nov 5. doi:10.1080/21645515.2019.168908.
- Ashley CW, Baecher-Allan C. Cutting edge: responder T cells regulate human DR+ effector regulatory T cell activity via granzyme B. J Immunol. 2009;183(8):4843–47. doi:10.4049/jimmunol.0900845.
- Cerignoli F, Abassi YA, Lamarche BJ, Guenther G, Santa Ana D, Guimet D, Zhang W, Zhang J, Xi B. In vitro immunotherapy potency assays using real-time cell analysis. PLoS One. 2018;13(3):e0193498. doi:10.1371/journal.pone.0193498.
- Revill PA, Chisari FV, Block JM, Dandri M, Gehring AJ, Guo H, Hu J, Kramvis A, Lampertico P, Janssen HLA, et al. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol Hepatol. 2019;4(7):545–58. doi:10.1016/S2468-1253(19)30119-0.
- Al-Mahtab M, Akbar SM, Aguilar JC, Uddin MH, Khan MS and Rahman S. Therapeutic potential of a combined hepatitis B virus surface and core antigen vaccine in patients with chronic hepatitis B. Hepatol Int. 2013;7(4):981–89. doi:10.1007/s12072-013-9486-4.
- Wang Z, Zhu K, Bai W, Jia B, Hu H, Zhou D, Zhang X, Zhang X, Xie Y, Bourgine MM, et al. Adenoviral delivery of recombinant hepatitis B virus expressing foreign antigenic epitopes for immunotherapy of persistent viral infection. J Virol. 2014;88(5):3004–15. doi:10.1128/JVI.02756-13.
- Cova L. Present and future DNA vaccines for chronic hepatitis B treatment. Expert Opin Biol Ther. 2017;17(2):185–95. doi:10.1080/14712598.2017.1265940.
- Wooddell CI, Rozema DB, Hossbach M, John M, Hamilton HL, Chu Q, Hegge JO, Klein JJ, Wakefield DH, Oropeza CE, et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol Ther. 2013;21(5):973–85. doi:10.1038/mt.2013.31.
- Du K, Liu J, Broering R, Zhang X, Yang D, Dittmer U and Lu M. Recent advances in the discovery and development of TLR ligands as novel therapeutics for chronic HBV and HIV infections. Expert Opin Drug Discov. 2018;13(7):661–70. doi:10.1080/17460441.2018.1473372.
- Gao Y, Zhang TY, Yuan Q and Xia NS. Antibody-mediated immunotherapy against chronic hepatitis B virus infection. Hum Vaccin Immunother. 2017;13(8):1768–73. doi:10.1080/21645515.2017.1319021.
- Yang L, Lu M. Small molecule inhibitors of hepatitis B virus nucleocapsid assembly: a new approach to treat chronic HBV infection. Curr Med Chem. 2018;25(7):802–13. doi:10.2174/0929867324666170704121800.
- Akbar SM, Horiike N, Onji M and Hino O. Dendritic cells and chronic hepatitis virus carriers. Intervirology. 2001;44(4):199–208. doi:10.1159/000050047.
- Vigano M, Grossi G, Loglio A and Lampertico P. Treatment of hepatitis B: is there still a role for interferon? Liver Int. 2018;38(Suppl 1):79–83. doi:10.1111/liv.13635.
- Janssen HL, van Zonneveld M, Senturk H, Zeuzem S, Akarca US, Cakaloglu Y, Simon C, So TM, Gerken G, de Man RA, et al. Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: a randomised trial. Lancet. 2005;365(9454):123–29. doi:10.1016/S0140-6736(05)17701-0.
- Marcellin P, Lau GK, Bonino F, Farci P, Hadziyannis S, Jin R, Lu ZM, Piratvisuth T, Germanidis G, Yurdaydin C, et al. Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N Engl J Med. 2004;351(12):1206–17. doi:10.1056/NEJMoa040431.
- Lau GK, Piratvisuth T, Luo KX, Marcellin P, Thongsawat S, Cooksley G, Gane E, Fried MW, Chow WC, Paik SW, et al. Peginterferon Alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N Engl J Med. 2005;352(26):2682–95. doi:10.1056/NEJMoa043470.
- Piccolo P, Lenci I, Demelia L, Bandiera F, Piras MR, Antonucci G, Nosotti L, Mari T, De Santis A, Ponti ML, et al. A randomized controlled trial of pegylated interferon-alpha2a plus adefovir dipivoxil for hepatitis B e antigen-negative chronic hepatitis B. Antivir Ther. 2009;14(8):1165–74. doi:10.3851/IMP1466.
- Tangkijvanich P, Chittmittraprap S, Poovorawan K, Limothai U, Khlaiphuengsin A, Chuaypen N, Wisedopas N and Poovorawan Y. A randomized clinical trial of peginterferon alpha-2b with or without entecavir in patients with HBeAg-negative chronic hepatitis B: role of host and viral factors associated with treatment response. J Viral Hepat. 2016;23(6):427–38. doi:10.1111/jvh.12467.
- Marcellin P, Ahn SH, Ma X, Caruntu FA, Tak WY, Elkashab M, Chuang WL, Lim SG, Tabak F, Mehta R, et al. Combination of tenofovir disoproxil fumarate and peginterferon alpha-2a increases loss of hepatitis B surface antigen in patients with chronic hepatitis B. Gastroenterology. 2016;150(1):134–144.e10. doi:10.1053/j.gastro.2015.09.043.
- Ahn SH, Marcellin P, Ma X, Caruntu FA, Tak WY, Elkhashab M, Chuang WL, Tabak F, Mehta R, Petersen J, et al. Hepatitis B surface antigen loss with tenofovir disoproxil fumarate plus peginterferon Alfa-2a: week 120 analysis. Dig Dis Sci. 2018;63(12):3487–97. doi:10.1007/s10620-018-5251-9.
- Lampertico P, Brunetto MR, Craxi A, Gaeta GB, Rizzetto M, Rozzi A and Colombo M. Add-on peginterferon alfa-2a to nucleos(t)ide analogue therapy for caucasian patients with hepatitis B ‘e’ antigen-negative chronic hepatitis B genotype D. J Viral Hepat. 2019;26(1):118–25. doi:10.1111/jvh.12999.
- Belkaid Y, Rouse BT. Natural regulatory T cells in infectious disease. Nat Immunol. 2005;6(4):353–60. doi:10.1038/ni1181.
- Suvas S, Kumaraguru U, Pack CD, Lee S and Rouse BT. CD4+CD25+ T cells regulate virus-specific primary and memory CD8+ T cell responses. J Exp Med. 2003;198(6):889–901. doi:10.1084/jem.20030171.
- Franzese O, Kennedy PT, Gehring AJ, Gotto J, Williams R, Maini MK and Bertoletti A. Modulation of the CD8+-T-cell response by CD4+ CD25+ regulatory T cells in patients with hepatitis B virus infection. J Virol. 2005;79(6):3322–28. doi:10.1128/JVI.79.6.3322-3328.2005.
- Stoop JN, van der Molen RG, Baan CC, van der Laan LJ, Kuipers EJ, Kusters JG and Janssen HL. Regulatory T cells contribute to the impaired immune response in patients with chronic hepatitis B virus infection. Hepatology. 2005;41(4):771–78. doi:10.1002/hep.20649.
- Peng G, Li S, Wu W, Sun Z, Chen Y and Chen Z. Circulating CD4+ CD25+ regulatory T cells correlate with chronic hepatitis B infection. Immunology. 2008;123(1):57–65. doi:10.1111/j.1365-2567.2007.02691.x.
- Kosinska AD, Zhang E, Lu M, Roggendorf M. Therapeutic vaccination in chronic hepatitis B: preclinical studies in the woodchuck. Hepat Res Treat. 2010;2010:817580.
- Maini MK, Pallett LJ. Defective T-cell immunity in hepatitis B virus infection: why therapeutic vaccination needs a helping hand. Lancet Gastroenterol Hepatol. 2018;3(3):192–202. doi:10.1016/S2468-1253(18)30007-4.
- Maini MK, Schurich A. The molecular basis of the failed immune response in chronic HBV: therapeutic implications. J Hepatol. 2010;52(4):616–19. doi:10.1016/j.jhep.2009.12.017.
- Bertoletti A, Kennedy PT. The immune tolerant phase of chronic HBV infection: new perspectives on an old concept. Cell Mol Immunol. 2015;12(3):258–63. doi:10.1038/cmi.2014.79.
- Protzer U, Knolle P. “To be or not to be”: immune tolerance in chronic hepatitis B. Gastroenterology. 2016;151(5):805–06. doi:10.1053/j.gastro.2016.09.038.
- Leung NW, Lai CL, Chang TT, Guan R, Lee CM, Ng KY, Lim SG, Wu PC, Dent JC, Edmundson S, et al. Extended lamivudine treatment in patients with chronic hepatitis B enhances hepatitis B e antigen seroconversion rates: results after 3 years of therapy. Hepatology. 2001;33(6):1527–32. doi:10.1053/jhep.2001.25084.
- Li J, Bao M, Ge J, Ren S, Zhou T, Qi F, Pu X and Dou J. Research progress of therapeutic vaccines for treating chronic hepatitis B. Hum Vaccin Immunother. 2017;13(5):986–97. doi:10.1080/21645515.2016.1276125.
- TH K, CB K, Guo Z, Mann D, Lu Y, Coeshott C, AJ G, Bertoletti A, ZZ H, Delaney W, et al. A whole recombinant yeast-based therapeutic vaccine elicits HBV X, S and core specific T cells in mice and activates human T cells recognizing epitopes linked to viral clearance. PLoS One. 2014;9(7):e101904. doi:10.1371/journal.pone.0101904.
- Lok AS, Pan CQ, Han SH, Trinh HN, Fessel WJ, Rodell T, Massetto B, Lin L, Gaggar A, Subramanian GM, et al. Randomized phase II study of GS-4774 as a therapeutic vaccine in virally suppressed patients with chronic hepatitis B. J Hepatol. 2016;65(3):509–16. doi:10.1016/j.jhep.2016.05.016.
- Xu DZ, Wang XY, Shen XL, Gong GZ, Ren H, Guo LM, Sun AM, Xu M, Li LJ, Guo XH, et al. Results of a phase III clinical trial with an HBsAg-HBIG immunogenic complex therapeutic vaccine for chronic hepatitis B patients: experiences and findings. J Hepatol. 2013;59(3):450–56. doi:10.1016/j.jhep.2013.05.003.
- Zoulim F, Fournier C, Habersetzer F, Sprinzl M, Pol S, Coffin CS, Leroy V, Ma M, Wedemeyer H, Lohse AW, et al. Safety and immunogenicity of the therapeutic vaccine TG1050 in chronic hepatitis B patients: a phase 1b placebo-controlled trial. Hum Vaccin Immunother. 2019. pp.1–12. doi:10.1080/21645515.2019.1651141.
- Zaiss AK, Machado HB and Herschman HR. The influence of innate and pre-existing immunity on adenovirus therapy. J Cell Biochem. 2009;108(4):778–90. doi:10.1002/jcb.22328.
- Fausther-Bovendo H, Kobinger GP. Pre-existing immunity against Ad vectors: humoral, cellular, and innate response, what’s important? Hum Vaccin Immunother. 2014;10(10):2875–84. doi:10.4161/hv.29594.
- Buchbinder SP, Mehrotra DV, Duerr A, Fitzgerald DW, Mogg R, Li D, Gilbert PB, Lama JR, Marmor M, Del Rio C, et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the step study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372(9653):1881–93. doi:10.1016/S0140-6736(08)61591-3.
- Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang XF, Dorta-Estremera SM, Greeley NR, Nitti G, Peng W, et al. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med. 2013;19(4):465–72. doi:10.1038/nm.3105.
- Shortman K, Lahoud MH and Caminschi I. Improving vaccines by targeting antigens to dendritic cells. Exp Mol Med. 2009;41(2):61–66. doi:10.3858/emm.2009.41.2.008.
- Wang XY, Wang B and Wen YM. From therapeutic antibodies to immune complex vaccines. NPJ Vaccines. 2019;4:2. doi:10.1038/s41541-018-0095-z.
- Gall VA, Philips AV, Qiao N, Clise-Dwyer K, Perakis AA, Zhang M, Clifton GT, Sukhumalchandra P, Ma Q, Reddy SM, et al. Trastuzumab increases HER2 uptake and cross-presentation by dendritic cells. Cancer Res. 2017;77(19):5374–83. doi:10.1158/0008-5472.CAN-16-2774.
- Wen YM, Mu L and Shi Y. Immunoregulatory functions of immune complexes in vaccine and therapy. EMBO Mol Med. 2016;8(10):1120–33. doi:10.15252/emmm.201606593.
- Weiner LM, Surana R and Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10(5):317–27. doi:10.1038/nri2744.
- Salazar G, Zhang N, Fu TM and An Z. Antibody therapies for the prevention and treatment of viral infections. NPJ Vaccines. 2017;2:19. doi:10.1038/s41541-017-0019-3.
- Delamarre L, Pack M, Chang H, Mellman I and Trombetta ES. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science. 2005;307(5715):1630–34. doi:10.1126/science.1108003.
- Manca F, Fenoglio D, Li Pira G, Kunkl A and Celada F. Effect of antigen/antibody ratio on macrophage uptake, processing, and presentation to T cells of antigen complexed with polyclonal antibodies. J Exp Med. 1991;173(1):37–48. doi:10.1084/jem.173.1.37.
- Noujaim AA, Schultes BC, Baum RP, Madiyalakan R. Induction of CA125-specific B and T cell responses in patients injected with MAb-B43.13–evidence for antibody-mediated antigen-processing and presentation of CA125 in vivo. Cancer Biother Radiopharm. 2001;16(3):187–203. doi:10.1089/10849780152389384.