
ABSTRACT
Glycosylation is an important post-translational modification, giving rise to a diverse and abundant repertoire of glycans on the cell surface, collectively known as the glycome. When focusing on immunity, glycans are indispensable in virtually all signaling and cell-cell interactions. More specifically, glycans have been shown to regulate key pathophysiological steps within T cell biology such as T cell development, thymocyte selection, T cell activity and signaling as well as T cell differentiation and proliferation. They are of major importance in determining the interaction of human T cells with tumor cells. In this review, we will describe the role of glycosylation of human T cells in more depth, elaborate on the importance of glycosylation in the interaction of human T cells with tumor cells and discuss the potential of cancer immunotherapies that are based on manipulating the glycome functions at the tumor immune interface.Citation1,Citation2
Introduction
Mammalian glycosylation
Glycosylation is the enzymatic process that leads to the formation of glycosidic linkages between carbohydrates and other carbohydrates, proteins or lipids. Next to proteolytic cleavage and modification of disulfide bonds, glycosylation is an important post-translational modification that gives rise to a diverse and abundant repertoire of glycans on the cell surface, collectively known as the glycome.1–Citation3 Known human glycans are formed almost exclusively from only ten monosaccharide building blocks (L-fucose, D-galactose, N-acetyl-D-galactosamine (GalNAc), N-acetyl-D-glucosamine (GlcNAc), D-glucuronic acid, D-iduronic acid, D-mannose, N-acetylneuraminic acid and D-xylose), which can be modified in particular glycoconjugates with sulfation, phosphorylation or de-acetylation. Through the orchestrated action of a diverse set of glycosyltransferases, glycosidases, glycan phosphorylases and polysaccharide lyases, a large diversity of mammalian glycans is generated. The biosynthesis of glycans is primarily determined by glycosyltransferases, enzymes that use activated sugar nucleotides or dolichol-phosphate bound donors as substrate in the enzymatic formation of glycosidic linkages to initiate, extend and diversify glycan structures. The expression of these glycosyltransferases is often highly regulated in a cell-type specific manner that changes during development, differentiation and activation, implying that changes in the glycome can occur in response to environmental and genetic stimuli.Citation4 However, the exact regulation of glycan synthesis is still not completely understood, and this is mainly due to the complexity inherent to the macromolecule itself and its nontemplated mode of biosynthesis.Citation5 The latter means that genomic information is not just ‘decoded’ by the ribosome translating the linear DNA code to a linear product, but that genomic information is ‘translated’ by a combination of transcriptional and membrane transport processes into a specific Golgi configuration.Citation6
The most frequently occurring types of mammalian protein glycosylation are N- and mucin type O-linked glycosylation and they lead to a large structural repertoire of glycans present on nearly every secreted and integral membrane protein. N-glycans are relatively bulky structures of which the synthesis starts on a lipid precursor at the cytoplasmic face of the endoplasmic reticulum (ER). After transfer to the ER lumen by an ER-resident flippase and further addition of monosaccharides, the glycan is transferred to nascent polypeptides at the side-chain amide nitrogen on asparagine residues in an N-x-S/T consensus sequence. After playing its role in protein folding quality control, the glycan is trimmed to a high mannose type structure carrying terminal mannoses. After transport from the ER to the Golgi apparatus (GA), the glycan can be further trimmed. At this point, these oligomannose glycans can remain unchanged, but most are subsequently rebuilt into hybrid and complex type glycans by the sequential action of GA resident glycosyltransferases. Typically, N-glycans develop into structures with at least two branches.
In contrast to N-glycans, O-glycans and protein-linked glycosaminoglycans (proteoglycans) are de novo synthesized, directly on the folded protein. This process is initiated in the GA, by the addition of D-GalNAc (mucin type O-glycans) or D-xylose (proteoglycans) to the side-chain hydroxyl group of serine or threonine. Subsequently, the glycans are turned into mature structures by the sequential action of a host of Golgi-resident enzymes. For mucin type O-glycans, this leads to a variety of core structures differing in their carbohydrate composition and linkage to the protein-proximal GalNAc residue, which are further extended and capped with similar structures as for N-glycans. ()
Figure 1. Overview of human N- and O-glycosylation in the Golgi apparatus. On the left side, the synthesis of a human glycoprotein with several relevant complex-type N-glycans is shown. In the cis Golgi, mannosidase I (ManI) activity leads to a Man5GlcNAc2 that can be further modified in the medial Golgi. N-acetylglucosaminyltransferase I (GnTI) activity commits the glycan to the complex or hybrid type. Mannosidase II (ManII) activity, followed by several N-acetylglucosaminyltransferases then further commits the glycan to the complex type. If only N-acetylglucosaminyltransferases II (GnTII) acts on it, the result is a biantennary complex type N-glycan. GnTIV and/or GnTV activity then generates different triantennary or a tetraantennary complex type glycan. Fucosyltransferase VIII (FucTVIII) can act on any complex or hybrid type glycan to add a core α-1,6-fucose in the medial Golgi. Afterward, in the trans Golgi, galactosyltransferases (GalT), fucosyltransferases (FucT), sialyltransferases (SiaT) or a combination of GnTs and GalTs synthesize different capping moieties (sialylation, poly-LacNAc repeats, Lewis antigens) on N-glycans
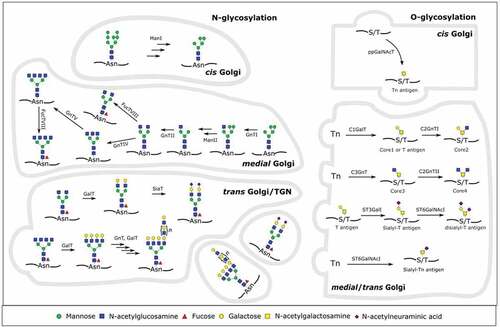
The glycans produced in the GA obviously depend on the repertoire of glycosylation enzymes present and their kinetic rate constants. Examination of transcripts encoding specific enzymatic machinery for glycan biosynthesis showed that many, but not all, changes in glycan abundance result from alterations in transcript expression of corresponding biosynthetic enzymes.Citation8,Citation9 This suggests that transcriptional regulation contributes significantly to the regulation of glycan expression.
Besides, systems glycobiology approaches have shown that additional factors and parameters of regulation should be considered, including processes regulating the localization of enzymes in the Golgi, the availability of substrates, enzyme kinetics ….Citation10,Citation11 The role of the GA in the translation of genome to glycome has been extensively reviewed in ref.Citation6 and will not further be discussed in this review. We can generally state that the relative abundance of glycans in the total glycan composition of a cell or tissue is in part dependent on the competition between glycosyltransferases with different activated donor substrate specificities for a certain glycoprotein acceptor. Furthermore, the concentration of sugar nucleotide donors, the association with specific chaperones, the modification or removal of sugars by additional enzymes and the trafficking dynamics of the acceptor glycoprotein throughout the secretory pathway are important factors contributing to the broad variety of glycans present on a cell.Citation6,Citation12
When focusing on immunity, glycans are indispensable in virtually all signaling and cell-cell interactions. Glycan signatures present on healthy, inflamed or malignant tissue, or pathogens, provide signals for “self” or “non-self” recognition and are increasingly appreciated as key molecules in regulating immunity versus tolerance.
For example, during the initiation of innate immune responses, the microbial molecular patterns that are recognized often are glycoconjugates. In adaptive immunity, glycans are involved in the alignment of the immunological synapse, in the generation and loading of antigenic peptides into MHC class I and in MHC class II antigen processing. These effects of glycans on immunity are reviewed by Baum et al.Citation13 In this review, we will focus on the role of glycans in human T cell homeostasis and their function in T cell immunotherapy for cancer ().
Glycan-binding proteins
In most cases, no single function can be attributed to a specific type of glycan. The function of a glycan is highly specific to the glycoprotein to which it is linked and the density at which it is present on a protein or on a biological surface, and it often relies upon the binding to glycan-binding proteins (GBPs). GBPs are characterized by one or more carbohydrate recognition domains and are often oligomeric or membrane bound. As the interactions between monosaccharides and proteins are often intrinsically weak, GBPs engage in multivalent interactions in order to confer avidity and specificity to their binding interactions with larger glycan structures or glycan-carrying surfaces, while their interactions are still tunable by alterations in the density of the glyco-epitopes to which they bind. GBPs are present in all organisms, ranging from microbes to humans and are involved in diverse biological processes, including immune responses. It will become clear from the following paragraphs that a particular glycan can be bound by several GBPs, and thus exerting pleiotropic effects. Since we will focus on the role of glycans and GBPs in human T cell function, we will first briefly introduce three types of GBPs with important roles in T cell responses.
Galectins
Galectins are a type of soluble, mammalian glycan-binding proteins grouped because of their common carbohydrate recognition domain (CRD) structure. It is because of variations in the amino acid sequence of this CRD that distinct Galectins have different glycan ligand specificities. As such, 16 different Galectins can be discerned.Citation14 N-acetyllactosamine is the ligand recognized by Galectins on the glycan moiety on both glycolipids and glycoproteins. The affinity of these interactions is proportional to the N-acetyllactosamine content of the glycoprotein or -lipid and the fine glycan structure. The spatial organization and modifications by glycosyltransferases are also contributing to the specificity of the interaction. For example, incorporation of fucose slightly improves Galectin-1 and Galectin-3 binding.Citation15
Galectins exert a broad range of effects during all aspects of T cell mediated immunity by the formation of lattices on the T cell surface.Citation16 In this way, they serve both to amplify or resolve immune responses. The precise effect varies depending on the tissue context, the intracellular or extracellular localization of the Galectin, the (patho)physiological condition and the presence of other regulatory mechanisms. Galectin-1, −3 and −9 are the main Galectins affecting T cell function, as will be discussed further below.
Selectins
Selectins are a subtype of C-type Lectin receptors (CLRs), membrane anchored GBPs which bind to sialylated, fucosylated or sulfated glycans, mainly on the O-glycan chains of glycoproteins or proteoglycans and on glycolipids. Selectins can be further subdivided into E-, P- and L-Selectin based on their expression profile and the glycan ligands to which they bind. While during inflammation, L-Selectin expression is induced in leukocytes, the expression of E-Selectin is induced on the vascular endothelium, and P-Selectin expression or translocation is induced on both vascular endothelium and platelets respectively. E- and P-Selectin bind sLex antigen (sialylated, fucosylated lactosamine core structures – sLex). The major glycoprotein ligand for P-Selectin, termed P-Selectin glycoprotein ligand-1 (PSGL-1), has sulfated tyrosine residues adjacent to a core-2-based O-glycan expressing SLex.Citation17 Ligands for L-Selectin contain 6-sulfo-sLex moieties on mucin-type O-glycans and N-glycans. Synthesis of the Selectin ligands mainly depends on the expression of the relevant fucosyl- and sialyltransferases. In the absence of these enzymes, lymphocyte homing to peripheral lymph nodes or sites of inflammation is strongly attenuated.Citation18,Citation19
Siglecs
Siglecs (sialic acid-binding immunoglobulin-type lectins) are a family of immune regulatory receptors predominantly found on the cells of the hematopoietic system. They have different binding preferences for sialylated glycan ligands. Siglecs are categorized into two subgroups that are functionally diverse, even in terms of signaling mechanisms. One group includes the structurally conserved Siglecs consisting of Siglec-1 (Sialoadhesin), Siglec-2 (CD22), Siglec-4 (myelin-associated glycoprotein, MAG) and Siglec-15. Another subgroup includes Siglec 3/CD33 and CD33-related Siglecs which have high homology to CD33 in their extracellular domains, contain at least one classic immunoreceptor tyrosine-based inhibition motif (ITIM) and show high variability in number between species and even vary between individuals of the same species, e.g. in humans. There is no evidence for a significant degree of functional redundancy either in the Siglecs conserved across species or in the CD33-related group of Siglecs. Most Siglecs function as inhibitory receptors on innate and adaptive immune cells. Binding to and subsequent signaling by both cis-ligands (expressed on the same cells; considered markers of ‘self’) as well as to sialoglycoconjugates presented in trans (e.g. on a neighboring cell or pathogen) attenuates immune responses.Citation20
Glycosylation in human T cell homeostasis
Role of glycosylation in T cell development
Glycans regulate key physiological steps within T cell biology such as T cell development and thymocyte selection, T cell activity and signaling as well as T cell differentiation and proliferation ().
Table 1. Role of T cell glycosylation during T cell development, activation and signaling
T cell migration is thought to be largely dictated by O-glycan modifications on Selectin ligands.Citation29 Before they can populate peripheral tissues, hematopoietically derived T cell precursors exit the bone marrow and must home to the thymus to mature from the double negative (DN) state toward double positive (DP) and undergo positive and negative selection to mature into either a CD4+ or CD8+ T cell. This homing is dependent on P-Selectin expression on thymic endothelial cells and the ability of T cell precursors to generate P-Selectin ligands.Citation48 Trafficking of T cell precursors to the thymus is impaired in the absence of PSGL-1 glycan fucosylation (α-1,3) since the latter is required for binding to P-Selectin on thymic epithelium.Citation49
The early thymic progenitor cells are CD4 and CD8 DN, and are also T cell receptor (TCR) negative. DN1 cells can give rise to T cells, NK cells, B cells, and macrophages. TCR rearrangement begins in DN2 cells. Commitment to the T cell lineage occurs during the transition from the DN2 to the DN3 stage. DN3 cells that have successfully rearranged their TCRβ-chain, form a pre-TCR complex with a pre-TCRα chain. This pre-TCR enforces β-selection and supports proliferation and differentiation bringing cells from DN3 to DN4. DN4 cells upregulate CD8 and CD4 to become DP and initiate rearrangement of the TCRα locus. After successful rearrangement, a TCRαβ complex is expressed on DPs that undergo positive or negative selection and become MHC class I or II restricted. This process upregulates glucose and glutamine uptake with a concomitant increase in O-GlcNAc levels.Citation21
O-GlcNAcylation has been shown to be required throughout T cell development.Citation50
Mice that are conditionally knockout for Ogt in DN thymocytes, contain substantially less thymocytes and mature CD4+ and CD8+ T cells, a phenotype consistent with a loss of β-selection.Citation21 Deletion of Ogt just before the DP stage, causes failure to differentiate to mature single positive (SP) CD4+ or CD8+ T cells, although numbers of DP cells are not influenced.Citation21 Increased expression of OGT in T cells from women with active lupus highlights the importance of O-GlcNAc regulation for normal immune homeostasis.Citation22
During T cell maturation from DP thymocytes into SP T cells, both α(−2,3)- and α(−2,6)-sialylation of cell surface glycoproteins is increased, as is experimentally shown by increased Sambucus nigra lectin (SNA) binding (specific for α(−2,6)-sialylation) and decreased peanut agglutinin (PNA) binding (specific for non-sialylated core-1 O-glycans).Citation23,Citation24 These findings are confirmed in β-Galactoside-α-2,6-Sialyltransferase 1 (ST6 GalI)-deficient mice, where DN populations are reduced, whereas a reduction in mature CD8+ SP thymocytes is demonstrated in ST3 GalI-deficient mice (reduced sialylation of core 1 O-linked glycans).Citation25
Following their development and exit from the thymus, naive T cells enter the periphery where they continually survey the spleen and secondary lymphoid organs for an encounter with cognate antigen. Increased sialic-acid modifications of glycans on differentiated SP CD8+ thymic T cells decrease the binding avidity of CD8 for MHC I molecules, thereby regulating TCR affinity-dependent negative selection.Citation16,Citation26–28
Naive T cells express high levels of L-Selectin (CD62L) and are defined as being CD44lo/CD62Lhi in mice and CD45RA+/CD62Lhi in humans. Once a naive T cell is activated by antigen binding and co-stimulation, CD62L expression ceases and T cells become effector cells, most of them having a limited life span. Those that survive become long-lived memory T cells, which are characterized by 2 subsets, being central memory (TCM, CD62L+ CCR7+) or effector memory (TEM, CD62L− CCR7−) T cells. TCM actively survey lymph nodes due to the presence of L-Selectin, whereas TEM are limited to the circulation, spleen and non-lymphoid tissues due to its absence.
Naive T cells cannot synthesize core 2 O-glycans or bind to P (CD62P)- and E-Selectin (CD62E), which essentially excludes them from entering non-lymphoid tissues. Following stimulation of the T cell receptor, both CD8+ and CD4+ T cells increase expression of core 2 β-1,6-N-acetylglucosaminyltransferase-I (GCNT1), α-1,3-fucosyltransferase-VII (FUT7), and likely additional enzymes that facilitate core 2 O-glycan synthesis. This post-translational modification transforms surface proteins such as PSGL-1 and CD43 into P- and E-Selectin ligands to direct extravasation across activated vascular endothelium and entry into non-lymphoid tissues. Effector CD8+ T cells have increased binding to PNA, indicating lowered capping of core 1 O-glycans with α-2,3-linked sialic acids.Citation29,Citation30
Role of glycosylation in T cell activation
Glycosylation is also involved in the regulation of T cell activation and functioning, mainly by N-glycosylation of the TCR and co-receptors (). Glycans can act to stabilize complexes at the immunological synapses, serving as a protective coat for the underlying protein by inhibiting degradation by proteases. They can suppress aggregation of TCRs on the membrane, thereby reducing auto-activation.Citation51
Initiation of T cell responses depends on the recognition of an antigenic epitope presented in the context of a class I or class II MHC (MHCI/II) molecule. Glycans on both the TCRαβ chains and MHCII can impact T cell activation. N-glycans present on the TCR are thought to play a role in cell surface localization and the orientation and organization of the molecule itself rather than directly influencing MHC interactions.
β-1,6-N-acetylglucosaminyltransferase-V (MGAT5) is the enzyme responsible for the initiation of GlcNAc-β-(1,6)-branching on N-glycans and is involved in multiple aspects of T cell activation. β-(1,6)-N-glycan branching leads to an increase in N-acetyllactosamine modifications, the ligand of Galectins. It has been demonstrated that absence of Mgat5 and thus a decrease in N-acetyllactosamine, lowers T cell activation thresholds in vitro by enhancing TCR clustering due to the absence of Galectin-glycoprotein lattice formation.Citation31 This Galectin-mediated lattice is responsible for holding CD45 and the TCR signaling complex in close proximity via their O- and N-linked glycans (respectively) to prevent low-avidity T cell activation.Citation32 Along the same line, an increased incidence of autoimmune disease is seen in the absence of Mgat5 in vivo.Citation52 Furthermore, negative regulation of TCR signaling by β-1,6-GlcNAc-containing N-glycans promotes development of Th2 over Th1 responses, enhances Th2 polarization, and suggests a mechanism for the increased autoimmune disease susceptibility observed in Mgat5−/- mice.Citation33 On the other hand, Mgat5 expression can be induced by the anti–inflammatory cytokine IL-10, decreasing antigen sensitivity of CD8+ T cells during chronic infection.Citation53
N-glycans on MHCI are important for protein folding and trafficking to the cell surface and peptide loading of MHCI molecules.Citation37–40 MHCI molecules are heterodimers comprised of a polymorphic transmembrane heavy chain and non-glycosylated β2-microglobulin. Within the heavy chain, the single conserved site for N-glycosylation across all known alleles is Asn86. The relative homogeneity of the complex type N-glycans on human MHCI suggests that the glycan might play a role in differentiating self and non-self,Citation54 however, this possibility remains unexplored.
MHCII has three highly conserved N-glycosylation sites, two on the α-chain (Asn78 and Asn118) and one on the β-chain (Asn19). Glycosylation of MHCII is not required for proper folding or trafficking of MHCII and shows a great variety between different cell types (e.g.: DC vs. B cell vs. macrophage), such as differences in degree of terminal sialylation and branching patterns which can directly modulate glycopeptide binding and presentation,Citation41–44 while this has no impact on binding of unmodified peptide antigens. It is speculated that the glycans serve to extend the binding platform of MHCII to accommodate the larger glycoantigen molecules.
Glycosylation of transmembrane T cell surface proteins
CD43 and CD45 are the two most abundant glycoproteins present on the T cell surface and are expressed throughout all stages of T cell development, from DN thymocytes to memory T cells. CD43 and CD45 are decorated with O- and N-glycans, and these molecules function in T cell activation, differentiation and migration, T cell receptor signaling and T cell apoptosis.Citation55 Alternatively spliced isoforms of CD45 display different glycosylation sites. The expression of ST6GalI transferase has been proposed to modulate binding of Galectin-1 to CD45 on T cells;Citation45 α-2,6-sialylation of CD45 glycans caps the terminal Gal-β-1,4-GlcNAc-binding sites used as a ligand by Galectin-1, implying that reduced ST6GalI expression in activated T cells could lead to increased binding of Galectin-1, resulting in cell death. Furthermore, α-2,6-sialylation was shown to inhibit clustering of CD45 on T cells, leading to diminished signaling ().Citation45
CD25 is the high-affinity alpha subunit of the IL-2 receptor and surface expression and retention is modulated by N-glycan branching, thereby controlling T cell differentiation with impact on immune tolerance.Citation46 Abolishing complex and hybrid type N-glycosylation in favor of the oligomannose type on CD25 via Mgat1 deletion reduced surface expression and retention while upregulation of branching via GlcNac supplementation or Mgat5 overexpression had the opposite effect, by raising CD25 surface levels. Increased β-1,5-N-glycan branching on CD25 also skews IL-2 signaling toward iTreg differentiation over Th17 differentiation.Citation46 Furthermore, transcription factors both upstream (e.g. NFAT and NFκb),Citation56,Citation57 and downstream (cMyc) of IL-2 signaling require O-GlcNAc modifications for proper function.Citation21 For example, chemical inhibition of OGT in primary human T cells decreases production of IL-2 upon TCR stimulation. Lymph node homing of T cells and antigen-experienced DCs relies on the chemokine receptor CCR7 and its two ligands, CCL19 and CCL21. Leukocyte subsets express distinct patterns of sialylation of the human CCR7.Citation47 This sialylation provides steric hindrance for ligand binding, rendering the receptor less efficient at low chemokine concentrations. DCs secrete glycosidase that deglycosylates CCR7 on T cells, thereby enhancing receptor potency and increasing its signaling ability, while inhibiting receptor endocytosis.Citation47 Consistent with this finding is the observation that naive, recirculating T cells only express limited amounts of sialylated CCR7, whereas high levels are seen on activated T cells. Glycosylation of the receptor acts as a swinging door, providing on the one hand steric hindrance for chemokine binding and on the other hand, retaining bound chemokines within the binding pocket, keeping the receptor susceptible for desensitization.
Importance of glycosylation in the interaction of human T cells with tumor cells
One of the hallmarks of cancer biology is the altered tumor cell glycosylation characterized by enhanced branching and sialylation and incomplete glycan synthesis. Virtually all cancer types are associated with expression of common glycan epitopes such as Sia-Lex and Sia-Lea. Expression of these Siglec/Selectin ligands is correlated with enhanced metastasis and poor prognosis of cancer patients suffering from colon, gastric, prostate, renal, pancreatic or lung cancer.Citation58–60 Moreover, neo-antigens originate through glycosylation of tumor proteins, serving as targets for tumor-specific T cells.Citation61,Citation62 In this paragraph, we will highlight the importance of glycosylation in the interaction of human T cells with tumor cells, often contributing to the creation of T cell killing resistance pathways in cancer.
Immune checkpoints
It is well established that tumors are inherently immunosuppressive, both due to the expression of immune checkpoint molecules (e.g. programmed death ligand-1, PD-L1) and expression of immune-inhibitory molecules (e.g. anti–inflammatory cytokines and chemokines). Unlike CTLA-4 or programmed cell death protein-1 (PD-1), which are primarily expressed on immune cells, PD-L1 is expressed in cancer cells and macrophages and plays a major role in inhibiting the immune response. Binding of PD-L1 to its receptor, PD-1 on T cells, inhibits T cell proliferation, cytokine production and cytolytic activity. The majority of PD-L1 on human tumor tissues and cancer cell lines is glycosylated, which stabilizes the PD-L1 protein, thus strengthening inhibition of the immune response.Citation63,Citation64
Growth arrest of activated T cells is dependent on induction of the immune checkpoint cell surface cytotoxic T-lymphocyte-associated-antigen-4 (CTLA-4). CTLA-4 competes with the sequence-related receptor CD28 for CD80/86 ligand on antigen-presenting cells to positively (in the case of CD28) or negatively (in the case of CTLA-4) regulate T cell proliferation.Citation65 TCR activation signaling normally leads to increased Mgat5 activity and N-glycan branching. It was demonstrated that in Mgat5−/- T cells, the fractions of T cells expressing CTLA-4 on their cell surfaces, as well as the CTLA-4 expression levels are decreased following low levels of TCR stimulation while CD28 surface levels are comparable in Mgat5−/- vs WT T cells.Citation34 On the contrary, increased N-glycan branching on CTLA-4 molecules upon TCR activation is shown to enhance its retention at the T cell surface, thereby suppressing T cell activation and promoting immune tolerance ().Citation35,Citation36
Tim-3 is an immune checkpoint receptor that shares a similar expression pattern as PD-1 on T cells. Tim-3 is a coinhibitory receptor, meaning that ligand binding leads to inhibition of T cell proliferation and cytokine production. T cell mediated immunity is further regulated through the interaction of Tim-3 with Galectin-9 (see further).Citation66
Galectins
Galectin-9 is one of the ligands of Tim-3 and negatively regulates T cell immunity.Citation67 A recent study shows that the interaction between Tim-3, expressed on T cells and Galectin-9, expressed in the tumor environment, is implicated in the resistance to PD-1 blockade in cancer patients ().Citation78 Furthermore, increased serum levels of Galectin-9 and Tim-3 were observed in the plasma of patients with acute myeloid leukemia and chronic lymphocytic leukemia, turning Galectin-9 into a potential target in leukemia.Citation79–81
Table 2. Role of glycan binding proteins in T cell cancer immunity
Galectin-1 functioning is involved in many signaling pathways leading to anti–inflammatory activities by targeting multiple types of immune cells.Citation68 In T cells, it is controlling T cell effector function homeostasis by regulating activation, differentiation, survival and cytokine production.Citation69 Galectin-1 is known to be a pro-tumorigenic and proangiogenic factor in cancer progression and has been associated with immune disorders such as HIV.Citation82 Overexpression of Galectin-1 was observed to be linked to progression of many types of cancers, including osteosarcoma, breast, lung and prostate cancers and in melanoma. The secretion of Galectin-1 by tumors leads to tolerogenic signaling and immune suppression. Therefore, Galectin-1 is an emerging biomarker for the diagnosis, prognosis and treatment plan of several cancers and interest is arising in targeting Galectin-1-glycan interactions in the attempt to overcome cancer mediated immunosuppression ().Citation83
Galectin-3 has the unique feature of being able to crosslink its binding partners through the presence of an oligomerization domain. Binding of Galectin-3 to glycoproteins has both pro- and anti-apoptotic effects on T cells, dependent on its localization. Intracellular Galectin-3 blocks apoptosis by stabilizing the mitochondrial membrane and preventing cytochrome c release,Citation70 while extracellular Galectin-3 binds to glycoproteins such as CTLA-4 and Lag3 on the T cell surface, leading to inhibition and cell death of activated T cells.Citation71,Citation72 Endogenous Galectin-3 produced by activated T cells is recruited to the immunological synapse. There it negatively regulates T cell activation by destabilizing the immunological synapse through direct interactions with glycoproteins associated with the T cell receptor, and by promoting downregulation of the TCR.Citation84,Citation85 Another interesting finding is that binding of Galectin-3 to antigen-specific activated CD8βT cells inhibits their effector function within the tumor microenvironment ().Citation71
Selectins
Selectins play important roles during different stages of cancer progression, including recruitment of leukocytes to the tumor site, immune evasion mechanisms, dissemination and extravasation. As L-Selectin is believed to be involved in the acquisition of T cell effector function, it confers protective antitumor immunity. Consistent with this, accumulation of myeloid derived suppressor cells (MDSCs) was observed in cancer patients and is known to suppress both the innate and adaptive antitumor response through processes comprising downregulation of L-Selectin.Citation73 In contrast, L-Selectin present on the surface of leukemia cells promotes cancer progression in CLL patients by affecting their trafficking and distribution abilities ().Citation74
Siglecs
Siglec-15 is broadly upregulated on human cancer cells and tumor-infiltrating myeloid cells, directly suppressing antigen-specific T cell responses in vitro and in vivo.Citation77 Tumor-infiltrating CD11b+ cells from Siglec-15 KO mice significantly promoted CD8+ T cell proliferation as well as cytokine secretion compared to those from wild type mice ().Citation77
Siglecs are expressed at very low levels on normal T cells. Several recent reports demonstrate the expression of Siglec-9 on tumor-infiltrating cytotoxic CD8+ T cells in multiple types of cancers (colorectal, ovarian and non-small cell lung cancer (NSCLC), and melanoma).Citation75,Citation76 These Siglec-9-expressing tumor-infiltrating lymphocytes (TILs) as well as Siglec-9+ CD8+ T cells, in lower numbers in the peripheral blood of melanoma patients and healthy donors, exhibited an effector memory phenotype. Co-expression with several known inhibitory T-cell receptors, e.g. PD-1, CTLA-4 and TIM-3, is seen. Despite this inhibitory phenotype, functional responses of Siglec-9+ CD8+ T cells to CD3/C28 costimulation are higher than those observed for their Siglec-9− counterparts in vitro ().Citation76
A note of caution
While all of the studies done have provided valuable insight on the potential modulatory function of altered glycosylation in T cell biology, it should be noted that many conclusions have been drawn from rather simplified in vitro cellular immunology assay systems. To what extend these insights and alterations of glycans and their interactions with glycan binding proteins have an impact in realistic in vivo models of disease and in patients often remains to be established. The manipulations of the surface glycome of T cells and/or tumor cells affect the entire glycome in most cases, not restricted to one particular glycosylated molecule. Hence, the overall effect of such glycome engineering should still be considered highly unpredictable and worthy of exciting further research.
Targeting the tumor or T cell glycome for therapeutic purposes
Cancer immunotherapies targeting the interaction of human T cells with tumor cells by use of agents that bind to glycans or glycan binding proteins
Many studies have aimed to induce specific immune responses against tumor-associated glycans and glycoproteins.Citation86 For example, a high anti-glycan antibody response could be driven by a combination of adjuvants and an optimization of T cell help through the use of repetitive immunizations with a glycopeptide-displaying particle resulting in the elicitation of anti-glycopeptide-specific T cells.Citation86 In another glycan-enhanced tumor vaccination approach, the coupling of glycans such as Lewis antigens to tumor antigens could facilitate internalization of the antigen by DCs due to improved recognition by DC-SIGN. This in turn favors antigen cross presentation and stimulation of tumor-specific T cell responses.Citation87 Recently, a carbohydrate-based vaccine was developed for metastatic breast cancer targeting the glycolipid Globo-H. The vaccine induced antibodies to target not only Globo-H but also SSEA3 and SSEA4. Together, these three targeted glycolipids were found to be uniquely expressed not only on the cell surface of breast cancer but on 15 additional cancer types, broadening the application of this vaccine, which is currently tested in a phase I clinical trial (NCT02310464).Citation88,Citation89
In a different approach, cell-based immunotherapy utilizes living cells to harness the body’s natural immune system to fight disease. Chimeric antigen receptors (CARs) have been developed that recognize cancer-associated glycan epitopes of glycolipids and glycoproteins, such as the Lewis y antigen (Ley), the sialyl Tn O-glycan epitope (TAG72), disialoganglioside GD2 and a Tn glycoform of MUC-1.Citation90
Interfering with the interaction of surface glycans and their lectin receptors, for example by adding antibodies that block tumor-associated glycan-lectin interactions is yet another strategy which is explored as antitumor therapy and several applications look very promising and/or are already used in the clinic.Citation64,Citation91–94
Considering the abundance of Siglec ligands on the surface of tumor cells and the inhibitory nature of many Siglecs, multiple studies have recently been initiated, investigating the effect of altering the levels of sialylation on tumor cells.Citation95 They are targeted using blocking antibodies, aiming to improve cancer immunotherapy.Citation96 Anti-Siglec-15 monoclonal antibody (targeting tumor-infiltrated macrophages-TIMs) inhibited the growth of established tumors in murine models.Citation77 A clinical trial is ongoing to test the effect of an anti-human Siglec-15 mAB (NC318) in solid tumors, with promising very early results recently reported.Citation97 Worth noting is that Siglec-15 KO mice do not develop obvious physical abnormalities, which is consistent with rare expression on normal tissues and suggests minimal adverse effects for Siglec-15 blockade therapy. Recently, also Siglec-9 was considered as interesting target for cancer immunotherapy.Citation75,Citation76 Targeting immunologic checkpoints that include dominant regulatory circuits confined to the tumor microenvironment, such as Siglec-9/-15 receptor-ligand interactions, allow to selectively unleash the restricted repertoire of TILs and TIMs, while reducing the potential for uncontrolled T cell activation and associated immune-related adverse events.Citation98
Interfering with Selectin and Galectin functioning can also influence the interaction of T cells with tumor cells.Citation95,Citation99 For example, interference with L-Selectin-mediated trafficking by treating patients with idelalisib, a phosphoinositide-3-kinase δ inhibitor, in high endothelial venules could limit dissemination of CLL to lymph nodes.Citation74 Galectin inhibitors have been evaluated in combination with chemotherapy drugs (5-fluorouracil) against solid tumors in several clinical trials, however, only one trial has been completed so far, suggesting that not all Galectin inhibitors are effective.Citation100 Given the pleiotropic effect of galectins, there may be a narrow safe dosage window, although data in humans remains scarce. Immunotherapy using monoclonal antibodies blocking immune checkpoint molecules has been combined with Galectin inhibitors to enhance the therapeutic effect in one study.Citation100 TIM-3 inhibitors have shown similar efficacy as that of PD-1 inhibitors in preclinical research and many clinical trials are now focusing on the use of anti-TIM3 blocking antibodies.Citation101
Glycan-engineering and implications in immune therapy
To date, the precise experimental adaptation of glycan structures on a specific protein in a living cell is most often not possible, except for the removal or addition of glycosylation sites. Therefore, instead of focusing on specific glycoproteins, a systems approach is taken in which the structure of glycans on the cell surface as a whole is perturbed. This allows to link changes in overall cellular glycosylation with cellular fates and functions, but often makes it more difficult to dissect the precise molecular mechanisms underlying glycosylation-mediated signaling events and the downstream cellular consequences. Alteration of cellular glycosylation capacity has an impact on multiple cell surface receptors, their biogenesis, biophysical behavior and signal transduction. What is measured is the integrated result of all of these alterations on cellular behavior. But of course, it is cellular behavior that matters in the end for therapeutic effects. The ability to remodel glycans on the cell surface could hence lead to novel approaches to manipulate cellular physiology and functional behavior.
The potential of glycosylation engineering to enhance therapeutics has been investigated and utilized extensively in the field of protein biopharmaceuticals, for example in anti-cancer antibodies.Citation102,Citation103 Cell-based therapies are emerging fast in the treatment of diverse diseases, including autoimmune diseases and cancer. In order to enhance the function of therapeutic cells, extensive studies are being performed seeking for improved survival, proliferation and differentiation of these cells. In the following sections, we will describe glycodesign strategies used to endow therapeutic cells with new favorable properties and functions.
Genetic engineering approaches
Genetic engineering via gene knockdown, knockout, overexpression, knock in or selective nucleotide mutations can be used to target glycosylation enzymes or complete pathways in order to reduce or silence undesirable glycosyltransferase activities or to enhance certain aspects of glycosylation.
These genetic engineering approaches are based on developments in the field of genome engineering.Citation104–106 Although these techniques are very versatile and give us the opportunity to make permanent cellular glycan modifications, it has to be kept in mind that potential off-target effects, inefficient delivery systems, confounding epigenetic regulation of glycosylation pathways, compensatory glycan alterations and unpredictable alterations in cellular physiology can have a detrimental impact on the intended results. Fast developments in the genome editing field result in continually improving and newly emerging tools, which should eliminate several of these problems in the coming years.Citation107
Some interesting results were already obtained using genetic glycoengineering. For example, selectively removing conserved N-glycosylation sites in the constant regions of TCRα and β-chains increased the functional avidity (reflected by increased cytokine secretion, lytic capacity and tumor cell recognition) of T cells transduced with modified TCRs. This is caused by an improved αβTCR multimerization and decreased TCR-MHC dissociation, ultimately increasing the recognition of tumor cells bearing the target antigen ().Citation108
Table 3. Examples of glycan-engineering and its implications in immune therapy
In another study, overexpression of rat ST6GalI in murine and human T cell lines resulted in increased α-2,6-sialylation of N-glycans, thereby shielding LacNAc residues from Galectin-1 binding and reducing Galectin-1 induced cell death of the T cells ().Citation45
Using a genome-wide loss-of-function CRISPR-Cas9 based screening method, Okada et al. identified FUT8 (the core fucosyltransferase) as a positive regulator of cell-surface expression of PD-1, a marker of T cell exhaustion. Inhibition of FUT8 by genetic ablation reduced cell surface expression of PD-1 and enhanced T cell activation and anti-tumor immune responses ().Citation109
Metabolic glycoengineering
Metabolic glycoengineering (MGE) involves the incorporation of non-natural glycan constituents. This type of engineering is often exploited in order to label glycans for the purpose of imaging, tracking and identifying glycan structures.Citation120 On the other hand, the remodeling of glycan structures on the cell surface can provide a powerful means to modulate cellular functions and responses. In practice, living cells or even entire organisms are supplemented with synthetic monosaccharide precursors that either alter the natural flux through a biosynthetic pathway or substitute natural metabolites with non-natural analogs. These metabolites are processed by the natural biosynthetic pathway, eventually leading to altered glycoforms, potentially containing non-natural chemical groups. This approach is rather convenient because the analog can be directly added to the cell culture medium during therapeutic cell cultivation. However, the large-scale synthesis of the required monosaccharide analogs can turn out to be very expensive to implement such glyco-engineering strategies in real therapeutic cell manufacturing.
Inhibiting the biosynthesis of Galectin-1-binding N-acetyllactosamine (LacNAc) glycans via systemic delivery of non-natural fluorinated carbohydrate analogs increases the number of infiltrating tumor-specific cytotoxic T cells and intratumoral IFN-γ expression in mice.Citation110 Alternatively, intratumoral injection of thiodigalactoside, a Galectin-binding variant of lactose with enhanced glycosidase stability, leads to an increase in tumor-infiltrating cytotoxic T cell numbers and reduced tumor growth ().Citation111 Furthermore, the use of fluorinated fucose and sialic acid analogs blocks protein fucosylation and sialylation, leading to suppression of Lewis-x (Lex) and sLex antigen biosynthesis.Citation112 This impairs cancer cell adhesion and migration in vitro and prevents metastasis formation in vivo in mice.Citation121 Additionally, sialic acid biosynthesis blockade enhances tumor cell – T cell interactions and enhances the killing of tumor cells by cytotoxic T cells ().Citation113 Further research is needed to unravel the mechanisms underlying the enhanced susceptibility of sialic acid-depleted tumor cells to killing by cytotoxic T cells. One hypothesis is that removal of negatively charged sialic acids could influence the biophysical interaction between tumor cells and T cells, for instance by affecting MHC I–TCR interactions and subsequent signaling events.Citation26,Citation122 In inflammatory bowel disease, metabolic supplementation of mucosal T cells with GlcNAc leads to enhancement of N-glycosylation branching on the TCR, thereby controlling the T cell immune response ().Citation114
In an alternative metabolic glycoengineering approach, monosaccharide analogs are used to introduce non-natural chemical moieties (such as ketones, azides, alkynes, thiols, …) in glycans, enabling bioorthogonal conjugation of small molecules such as toxins, drugs, imaging agents and polymers via click chemistry. Using this bioorthogonal click chemistry, a library of more than 60 different cell lines was generated with different sialic acid modifications, leading to dramatic increases in binding with Siglec family members. This enabled the study and steering of the complex interactions between sialic acid and their Siglec binding partners.Citation123 Whether any of this will be of relevance for safe enhanced cellular immunotherapy awaits further experimentation. Glycometabolic bioorthogonal chemistry can be utilized to establish a highly efficient viral transduction system for human primary T lymphocytes, thereby showing a great potential for clinical-engineered T-lymphocyte manufacturing.Citation124
In order to conclude this section, it is important to point out the complex and often-unpredictable interplay between metabolism, genetics and cell fate occurring during metabolic glycan engineering. Only further rigorous experimentation in therapy-relevant cell manufacturing conditions and suitable animal models of cancer (in particular using patient-derived human tumor cells) may reveal the concepts that are promising for translation into therapy.
Chemoenzymatic glycoengineering
Chemoenzymatic glycan labeling and modification is an emerging valuable tool to modify the glycan structures within a cell, and is complementary to metabolic glycan engineering. Instead of using the cell’s own glycan biosynthetic machinery to incorporate unnatural monosaccharides, chemoenzymatic glycoengineering utilizes a recombinant glycosyltransferase to transfer natural or unnatural monosaccharides equipped with a bioorthogonal chemical tag to glycoconjugates, thereby endowing them with novel structural characteristics and function.Citation125
As described in the previous section, imaging glycans in vivo is possible using bioorthogonal chemical reporter strategies in which cells or even complete organisms are supplemented with azide- or alkyne-tagged monosaccharide precursors. A limitation of this strategy is that each monosaccharide is normally incorporated into a multitude of different glycans, making it impossible to label higher order glycans uniquely by feeding the biosynthetic pathway with unnatural monosaccharides. Chemoenzymatic approaches were reported for the labeling of complete cell surface glycans containing LacNAc. In this approach, recombinant microbial α-(1,3)-fucosyltransferase treatment was used to label the LacNAc moieties with clickable fucose analogs, which can be used as biophysical probes for imaging or glycomics analysis. By probing LacNAc levels in this way, it was possible to differentiate lymphocytes exhibiting different activation states ().Citation115
Ex vivo enzymatic treatment of immune cells has led to promising results in the field of anti-graft versus host disease adoptive cell therapy. When regulatory T cells were treated with recombinantly produced Fucosyltransferase-VI (FUT6) and the GDP-Fucose donor substrate, an increase in sLex antigens was observed on their cell surface, leading to an improvement in trafficking, homing to the site of inflammation and engraftment of these regulatory T cells ().Citation117 Similarly, increased homing of CAR-T cells to the bone marrow was observed following the same ex–vivo treatment with FucTVI.Citation126 When regulatory T cells are enzymatically treated with PNGaseF to partially remove N-glycans expressed on the surface, their capacity to suppress early activation events in the spleen is substantially impaired, leading to proliferation of naive and memory T cells ().Citation118 Furthermore, pretreatment of cytotoxic T cells with either a Clostridium perfringens sialidase or an O-linked glycoprotein endopeptidase increased activation and cytolytic capacity in vitro, indicating that glycosylated surface proteins can hinder cytotoxic T cell activation and functioning ().Citation119 More recently, a single-step chemoenzymatic approach for the development of antibody-cell conjugates was described. In this approach, the therapeutic T cell surface glycocalyx is ex vivo modified at LacNAc moieties with tumor-targeting antibodies. This is made possible by the use of a GDP-fucose analog, conjugated by click chemistry at the C6 position to a full length antibody, using Helicobacter pylori α-1,3-fucosyltransferase. In this way, T cells obtain specific tumor targeting capacity and resistance to inhibitory signals produced by tumor cells.Citation116
Concluding remarks
Despite the clinical success of antibodies against immune checkpoints, only a subset of people exhibits sustained responses. This can be explained by the fact that anti-tumor immunity is influenced by a continuously evolving complex interplay of host, tumor and environmental factors governing the strength and timing of the anticancer response.
These factors combine to produce a ‘cancer-immune set point’, which can be seen as the equilibrium between the factors that promote or suppress anticancer immunity, that can predict responses to immunotherapy (for a review, see ref.Citation127). It is now clear that glycan structures and their binding proteins play very important roles in this.
Glyco-engineering of the tumor cell – immune cell interface thus provides exciting perspectives for improving immunotherapy in the future. Due to the complexity, species differences and pleiotropic effects of manipulating this interface, the predictability of such glyco-engineering outcomes by extrapolation from in vitro or simple animal model systems will likely be low. It will inspire exciting research in glyco-oncology in models that are even more faithful representations of human disease, for many years to come. The first drugs that target glyco-components of the tumor-immune interface are in full clinical development, and it is likely that many more will follow.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Author contributions
EDB and NF reviewed the literature, wrote the text and made the tables. LM and NC carefully revised and co-wrote the manuscript. LM made and EDB made .
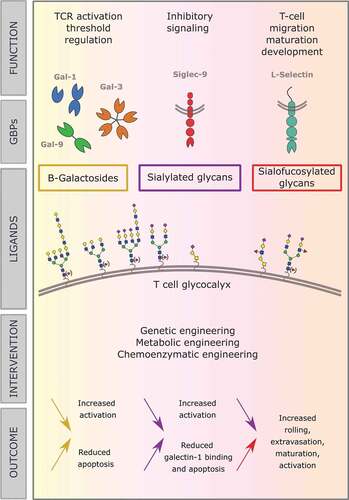
Figure 2. In this review, we discuss the role of different types of glycans decorating the surface of T cells, and their binding to GBPs and resulting functions. Central in the figure, several mammalian glycoforms that can occur on the T cell surface are shown. Above them, the GBPs that bind to them and the effects they have on T cells are outlined and below them we elaborate on the effects of therapeutic interventions on the glycan composition. The color of the arrows relates to the glycoforms, as indicated centrally in the figure, and the direction indicates decreased or increased expression of those glycoforms. The text next to the arrow then explains the effects on the T cell, resulting from the indicated changes in the glycoforms
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Abbreviations
| ADCC | = | antibody-dependent cellular cytotoxicity |
| Asn | = | asparagine |
| ASGPR | = | Asialoglycoprotein receptor |
| CAR | = | chimeric antigen receptor |
| CD62E | = | E-selectin |
| CD62L | = | L-selectin |
| CD62P | = | P-selectin |
| CLR | = | C-type Lectin receptor |
| CRD | = | carbohydrate recognition domains |
| CRISPR-CAS | = | Clustered Regularly Interspaced Short Palindromic Repeats |
| CTLA-4 | = | cytotoxic T-lymphocyte-associated antigen-4 |
| DC | = | dendritic cell |
| DC-SIGN | = | dendritic cell-specific ICAM-grabbing non-integrin |
| DN | = | double negative |
| DP | = | double positive |
| ER | = | endoplasmic reticulum |
| Fut6 | = | α1-3 fucosyltransferase VI |
| Fut7 | = | α1-3 fucosyltransferase VII |
| GA | = | Golgi apparatus |
| GalNac | = | N-acetylgalactosamine |
| GBP | = | glycan-binding protein |
| Gcnt1 | = | β1-6 N-acetylglucosaminyltransferase-I |
| GlcNAc | = | N-acetylglucosamine |
| HCV | = | hepatitis C virus |
| ITIM | = | immunoreceptor tyrosine-based inhibition motif |
| LacNac | = | N-acetyllactosamine |
| Lex | = | Lewis x antigen |
| Ley | = | Lewis y antigen |
| MAG | = | myelin-associated glycoprotein |
| MBP | = | mannose-binding protein |
| MDSC | = | myeloid derived suppressor cell |
| Mgat5 | = | β1,6 N-acetylglucosaminyltransferase V |
| MGE | = | metabolic glycoengineering |
| MHC | = | major histocompatibility complex |
| MMR | = | macrophage mannose receptor |
| NSCLC | = | non-small cell lung cancer |
| OGT | = | O-GlcNAc transferase |
| PD-1 | = | programmed cell death protein-1 |
| PD-L1 | = | programmed death ligand-1 |
| PNA | = | peanut agglutinin |
| PSGL-1 | = | P-selectin glycoprotein ligand-1 |
| Siglec | = | sialic acid-binding immunoglobulin-type lectins |
| sLex | = | sialyl-Lewis-X |
| SNA | = | Sambucus nigra |
| SSEA | = | stage-specific embryonic antigen 3 |
| ST3Gal1 | = | β-Galactoside α2,3-Sialyltransferase 1 |
| ST6Gal1 | = | β-Galactoside α2,6-Sialyltransferase 1 |
| TALEN | = | Transcription Activator-Like Effector Nucleases |
| TCM | = | central memory T cell |
| TCR | = | T cell receptor |
| TEM | = | effector memory T cell |
| TIM | = | tumor-infiltrated macrophage |
| TIL | = | tumor-infiltrating lymphocyte |
| Treg | = | regulatory T cell |
Acknowledgments
This work was supported through a PhD fellowship to EDB from FWO, by VIB and UGhent institutional funding to NF, LM and NC. This work was supported by grant G050420N FWO Vlaanderen.
Related Research Data
References
- Cook KM, Hogg PJ. Post-translational control of protein function by disulfide bond cleavage. Antioxid Redox Signal. 2012;18:1987–2015. doi:10.1089/ars.2012.4807.
- Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Molecular cell biology. 4th ed. New York, NY: W. H. Freeman; 2000. Section 17.6, Post-Translational Modifications and Quality Control in the Rough ER. Available from: https://www.ncbi.nlm.nih.gov/books/NBK21741/
- Rogers LD, Overall CM. Proteolytic post-translational modification of proteins: proteomic tools and methodology. Mol Cell Proteomics MCP. 2013;12:3532–42. doi:10.1074/mcp.M113.031310.
- Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol. 2012;13:448–62. doi:10.1038/nrm3383.
- Neelamegham S, Mahal LK. Multi-level regulation of cellular glycosylation: from genes to transcript to enzyme to structure. Curr Opin Struct Biol. 2016;40:145–52. doi:10.1016/j.sbi.2016.09.013.
- Pothukuchi P, Agliarulo I, Russo D, Rizzo R, Russo F, Parashuraman S. Translation of genome to glycome: role of the Golgi apparatus. FEBS Lett. 2019;593:2390–411. doi:10.1002/feb2.v593.17.
- De Wachter C, Van Landuyt L, Callewaert N. Engineering of yeast glycoprotein expression. In: Advances in Biochemical Engineering/Biotechnology. Berlin, Heidelberg: Springer; 2018. p. 1–43. https://link.springer.com/chapter/10.1007/10_2018_69
- Nairn AV, York WS, Harris K, Hall EM, Pierce JM, Moremen KW. Regulation of glycan structures in animal tissues. J Biol Chem. 2008;283:17298–313. doi:10.1074/jbc.M801964200.
- Nairn AV, Aoki K, Dela Rosa M, Porterfield M, Lim J-M, Kulik M, Pierce JM, Wells L, Dalton S, Tiemeyer M, et al. Regulation of glycan structures in murine embryonic stem cells. J Biol Chem. 2012;287:37835–56. doi:10.1074/jbc.M112.405233.
- Spahn PN, Lewis NE. Systems glycobiology for glycoengineering. Curr Opin Biotechnol. 2014;30:218–24. doi:10.1016/j.copbio.2014.08.004.
- Liu G, Neelamegham S. Integration of systems glycobiology with bioinformatics toolboxes, glycoinformatics resources, and glycoproteomics data. Wiley Interdiscip Rev Syst Biol Med. 2015;7:163–81. doi:10.1002/wsbm.2015.7.issue-4.
- Varki A. Biological roles of glycans. Glycobiology. 2017;27:3–49.
- Baum LG, Cobb BA. The direct and indirect effects of glycans on immune function. Glycobiology. 2017;27:619–24. doi:10.1093/glycob/cwx036.
- Thiemann S, Baum LG. Galectins and immune responses-just how do they do those things they do? Annu Rev Immunol. 2016;34:243–64. doi:10.1146/annurev-immunol-041015-055402.
- Horlacher T, Oberli MA, Werz DB, Kröck L, Bufali S, Mishra R, Sobek J, Simons K, Hirashima M, Niki T, et al. Determination of carbohydrate-binding preferences of human galectins with carbohydrate microarrays. Chembiochem Eur J Chem Biol. 2010;11:1563–73. doi:10.1002/cbic.v11:11.
- Starr TK, Daniels MA, Lucido MM, Jameson SC, Hogquist KA. Thymocyte sensitivity and supramolecular activation cluster formation are developmentally regulated: a partial role for sialylation. J Immunol. 2003;171:4512–20. doi:10.4049/jimmunol.171.9.4512.
- Cummings RD. Structure and function of the selectin ligand PSGL-1. Braz J Med Biol Res Rev Bras Pesqui Medicas E Biol. 1999;32:519–28. doi:10.1590/S0100-879X1999000500004.
- Homeister JW, Thall AD, Petryniak B, Malý P, Rogers CE, Smith PL, Kelly RJ, Gersten KM, Askari SW, Cheng G, et al. The alpha(1,3)fucosyltransferases FucT-IV and FucT-VII exert collaborative control over selectin-dependent leukocyte recruitment and lymphocyte homing. Immunity. 2001;15:115–26. doi:10.1016/S1074-7613(01)00166-2.
- Sperandio M, Frommhold D, Babushkina I, Ellies LG, Olson TS, Smith ML, Fritzsching B, Pauly E, Smith DF, Nobiling R, et al. Alpha 2,3-sialyltransferase-IV is essential for L-selectin ligand function in inflammation. Eur J Immunol. 2006;36:3207–15. doi:10.1002/eji.200636157.
- Pillai S, Netravali IA, Cariappa A, Mattoo H. Siglecs and immune regulation. Annu Rev Immunol. 2012;30:357–92. doi:10.1146/annurev-immunol-020711-075018.
- Swamy M, Pathak S, Grzes KM, Damerow S, Sinclair LV, van Aalten DMF, Cantrell DA. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat Immunol. 2016;17:712–20. doi:10.1038/ni.3439.
- Hewagama A, Gorelik G, Patel D, Liyanarachchi P, McCune WJ, Somers E, Gonzalez-Rivera T, Strickland F, Richardson B. Overexpression of X-Linked genes in T cells from women with lupus. J Autoimmun. 2013;41:60–71. doi:10.1016/j.jaut.2012.12.006.
- Baum LG, Derbin K, Perillo NL, Wu T, Pang M, Uittenbogaart C. Characterization of terminal sialic acid linkages on human thymocytes. J Biol Chem. 1996;271:10793–99. doi:10.1074/jbc.271.18.10793.
- Martin LT, Marth JD, Varki A, Varki NM. Genetically altered mice with different sialyltransferase deficiencies show tissue-specific alterations in sialylation and sialic acid 9-O-acetylation. J Biol Chem. 2002;277:32930–38. doi:10.1074/jbc.M203362200.
- Moody AM, Chui D, Reche PA, Priatel JJ, Marth JD, Reinherz EL. Developmentally regulated glycosylation of the CD8αβ coreceptor stalk modulates ligand binding. Cell. 2001;107:501–12. doi:10.1016/S0092-8674(01)00577-3.
- Daniels MA, Devine L, Miller JD, Moser JM, Lukacher AE, Altman JD, Kavathas P, Hogquist KA, Jameson SC. CD8 binding to MHC class I molecules is influenced by T cell maturation and glycosylation. Immunity. 2001;15:1051–61. doi:10.1016/S1074-7613(01)00252-7.
- Moody AM, North SJ, Reinhold B, Dyken SJV, Rogers ME, Panico M, Dell A, Morris HR, Marth JD, Reinherz EL. Sialic acid capping of CD8β core 1-O-glycans controls thymocyte-major histocompatibility complex class I interaction. J Biol Chem. 2003;278:7240–46. doi:10.1074/jbc.M210468200.
- Pappu BP, Shrikant PA. Alteration of cell surface sialylation regulates antigen-induced naive CD8+ T cell responses. J Immunol. 2004;173:275–84. doi:10.4049/jimmunol.173.1.275.
- Hobbs SJ, Nolz J. Regulation of T cell trafficking by enzymatic synthesis of O-glycans. Front Immunol. 2017;8:600. doi:10.3389/fimmu.2017.00600.
- Aguilar AL, Gao Y, Hou X, Lauvau G, Yates JR, Wu P. Profiling of protein O-GlcNAcylation in murine CD8+ effector- and memory-like T cells. ACS Chem Biol. 2017;12:3031–38. doi:10.1021/acschembio.7b00869.
- Dennis JW, Lau KS, Demetriou M, Nabi IR. Adaptive regulation at the cell surface by N-Glycosylation. Traffic. 2009;10:1569–78. doi:10.1111/tra.2009.10.issue-11.
- Earl LA, Bi S, Baum LG. N- and O-Glycans Modulate Galectin-1 Binding, CD45 Signaling, and T Cell Death. J Biol Chem. 2010;285:2232–44. doi:10.1074/jbc.M109.066191.
- Morgan R, Gao G, Pawling J, Dennis JW, Demetriou M, Li B. N-acetylglucosaminyltransferase V (Mgat5)-mediated N-glycosylation negatively regulates Th1 cytokine production by T cells. J Immunol Baltim Md 1950. 2004;173:7200–08.
- Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M, Dennis JW. Complex N-Glycan Number and Degree of Branching Cooperate to Regulate Cell Proliferation and Differentiation. Cell. 2007;129:123–34. doi:10.1016/j.cell.2007.01.049.
- Grigorian A, Torossian S, Demetriou M. T-cell growth, cell surface organization, and the galectin glycoprotein lattice. Immunol Rev. 2009;230:232–46. doi:10.1111/imr.2009.230.issue-1.
- Chen H-L, Li CF, Grigorian A, Tian W, Demetriou M. T cell receptor signaling co-regulates multiple golgi genes to enhance N-glycan branching. J Biol Chem. 2009;284:32454–61. doi:10.1074/jbc.M109.023630.
- Zhang W, Wearsch PA, Zhu Y, Leonhardt RM, Cresswell P. A role for UDP-glucose glycoprotein glucosyltransferase in expression and quality control of MHC class I molecules. Proc Natl Acad Sci U S A. 2011;108:4956–61. doi:10.1073/pnas.1102527108.
- Wearsch PA, Peaper DR, Cresswell P. Essential glycan-dependent interactions optimize MHC class I peptide loading. Proc Natl Acad Sci. 2011;108:4950–55. doi:10.1073/pnas.1102524108.
- Ryan SO, Cobb BA. Host glycans and antigen presentation. Microbes Infect Inst Pasteur. 2012;14:894–903. doi:10.1016/j.micinf.2012.04.010.
- Ryan SO, Cobb BA. Roles for major histocompatibility complex glycosylation in immune function. Semin Immunopathol. 2012;34:425–41. doi:10.1007/s00281-012-0309-9.
- Swiedler SJ, Freed JH, Tarentino AL, Plummer TH, Hart GW. Oligosaccharide microheterogeneity of the murine major histocompatibility antigens. Reproducible site-specific patterns of sialylation and branching in asparagine-linked oligosaccharides. J Biol Chem. 1985;260:4046–54.
- Ryan SO, Bonomo JA, Zhao F, Cobb BA. MHCII glycosylation modulates Bacteroides fragilis carbohydrate antigen presentation. J Exp Med. 2011;208:1041–53. doi:10.1084/jem.20100508.
- Cobb BA, Kasper DL. Characteristics of carbohydrate antigen binding to the presentation protein HLA-DR. Glycobiology. 2008;18:707–18. doi:10.1093/glycob/cwn050.
- Johnson JL, Jones MB, Ryan SO, Cobb BA. The Regulatory Power of Glycans and their Binding Partners in Immunity. Trends Immunol. 2013;34:290–98. doi:10.1016/j.it.2013.01.006.
- Amano M, Galvan M, He J, Baum LG. The ST6Gal I Sialyltransferase selectively modifiesN-Glycans on CD45 to negatively regulate galectin-1-induced CD45 clustering, phosphatase modulation, and T cell death. J Biol Chem. 2003;278:7469–75. doi:10.1074/jbc.M209595200.
- Araujo L, Khim P, Mkhikian H, Mortales C-L, Demetriou M. Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. eLife. 2017;6. doi:10.7554/eLife.21330.
- Hauser MA, Kindinger I, Laufer JM, Späte A-K, Bucher D, Vanes SL, Krueger WA, Wittmann V, Legler DF. Distinct CCR7 glycosylation pattern shapes receptor signaling and endocytosis to modulate chemotactic responses. J Leukoc Biol. 2016;99:993–1007. doi:10.1189/jlb.2VMA0915-432RR.
- Rossi FMV, Corbel SY, Merzaban JS, Carlow DA, Gossens K, Duenas J, So L, Yi L, Ziltener HJ. Recruitment of adult thymic progenitors is regulated by P-selectin and its ligand PSGL-1. Nat Immunol. 2005;6:626–34. doi:10.1038/ni1203.
- Sultana DA, Zhang SL, Todd SP, Bhandoola A. Expression of functional PSGL-1 on hematopoietic progenitors is developmentally regulated. J Immunol Baltim Md 1950. 2012;188:4385–93.
- Li Y, Xie M, Men L, Du J. O-GlcNAcylation in immunity and inflammation: an intricate system (Review). Int J Mol Med. 2019;44:363–74. doi:10.3892/ijmm.2019.4238.
- Daniels MA, Hogquist KA, Jameson SC. Sweet “n” sour: the impact of differential glycosylation on T cell responses. Nat Immunol. 2002;3:903–10. doi:10.1038/ni1002-903.
- Demetriou M, Granovsky M, Quaggin S, Dennis JW. Negative regulation of T-cell activation and autoimmunity by Mgat5 N -glycosylation. Nature. 2001;409:733. doi:10.1038/35055582.
- Smith LK, Boukhaled GM, Condotta SA, Mazouz S, Guthmiller JJ, Vijay R, Butler NS, Bruneau J, Shoukry NH, Krawczyk CM, et al. Interleukin-10 Directly Inhibits CD8+ T Cell Function by Enhancing N-Glycan Branching to Decrease Antigen Sensitivity. Immunity. 2018;48:299–312.e5. doi:10.1016/j.immuni.2018.01.006.
- An HJ, Froehlich JW, Lebrilla CB. Determination of glycosylation sites and site-specific heterogeneity in glycoproteins. Curr Opin Chem Biol. 2009;13:421–26. doi:10.1016/j.cbpa.2009.07.022.
- Clark MC, Baum LG. T cells modulate glycans on CD43 and CD45 during development and activation, signal regulation, and survival. Ann N Y Acad Sci. 2012;1253:58–67. doi:10.1111/j.1749-6632.2011.06304.x.
- Golks A, Tran -T-T-T, Goetschy JF, Guerini D. Requirement for O-linked N-acetylglucosaminyltransferase in lymphocytes activation. Embo J. 2007;26:4368–79. doi:10.1038/sj.emboj.7601845.
- Ramakrishnan P, Clark PM, Mason DE, Peters EC, Hsieh-Wilson LC, Baltimore D. Activation of the Transcriptional Function of the NF-κB Protein c-Rel by O-GlcNAc Glycosylation. Sci Signal. 2013;6:ra75. doi:10.1126/scisignal.2004097.
- McEver RP. Selectin-carbohydrate interactions during inflammation and metastasis. Glycoconj J. 1997;14:585–91. doi:10.1023/A:1018584425879.
- Borsig L. Selectins in cancer immunity. Glycobiology. 2018;28:648–55. doi:10.1093/glycob/cwx105.
- Häuselmann I, Borsig L. Altered tumor-cell glycosylation promotes metastasis. Front Oncol. 2014;4:28. doi:10.3389/fonc.2014.00028.
- RodrÍguez E, Schetters STT, van Kooyk Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat Rev Immunol. 2018;18:204–11. doi:10.1038/nri.2018.3.
- Peixoto A, Relvas-Santos M, Azevedo R, Santos LL, Ferreira JA. Protein Glycosylation and Tumor Microenvironment Alterations Driving Cancer Hallmarks. Front Oncol. 2019;9:380–380. doi:10.3389/fonc.2019.00380.
- Li C-W, Lim S-O, Xia W, Lee -H-H, Chan L-C, Kuo C-W, Khoo K-H, Chang -S-S, Cha J-H, Kim T, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632. doi:10.1038/ncomms12632.
- Li C-W, Lim S-O, Chung EM, Kim Y-S, Park AH, Yao J, Cha J-H, Xia W, Chan L-C, Kim T, et al. Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell. 2018;33:187–201.e10. doi:10.1016/j.ccell.2018.01.009.
- Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol. 2001;1:220–28. doi:10.1038/35105024.
- Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, Zheng XX, Strom TB, Kuchroo VK. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–52. doi:10.1038/ni1271.
- Wang H, Kaur G, Sankin AI, Chen F, Guan F, Zang X. Immune checkpoint blockade and CAR-T cell therapy in hematologic malignancies. J Hematol OncolJ Hematol Oncol. 2019;12:59. doi:10.1186/s13045-019-0746-1.
- Sundblad V, Morosi LG, Geffner JR, Rabinovich GA. Galectin-1: A Jack-of-All-Trades in the Resolution of Acute and Chronic Inflammation. J Immunol. 2017;199:3721–30. doi:10.4049/jimmunol.1701172.
- Clemente T, Vieira NJ, Cerliani JP, Adrain C, Luthi A, Dominguez MR, Yon M, Barrence FC, Riul TB, Cummings RD, et al. Proteomic and functional analysis identifies galectin-1 as a novel regulatory component of the cytotoxic granule machinery. Cell Death Dis. 2017;8:e3176. doi:10.1038/cddis.2017.506.
- Yu F, Finley RL, Raz A, Kim H-RC. Galectin-3 translocates to the perinuclear membranes and inhibits cytochrome c release from the mitochondria. A role for synexin in galectin-3 translocation. J Biol Chem. 2002;277:15819–27. doi:10.1074/jbc.M200154200.
- Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, Jaffee E. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol Res. 2015;3:412–23. doi:10.1158/2326-6066.CIR-14-0150.
- Stillman BN, Hsu DK, Pang M, Brewer CF, Johnson P, Liu F-T, Baum LG. Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J Immunol Baltim Md 1950. 2006;176:778–89.
- Ostrand-Rosenberg S. Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother CII. 2010;59:1593–600. doi:10.1007/s00262-010-0855-8.
- Lafouresse F, Bellard E, Laurent C, Moussion C, J-J F, Ysebaert L, Girard J-P. L-selectin controls trafficking of chronic lymphocytic leukemia cells in lymph node high endothelial venules in vivo. Blood. 2015;126:1336–45. doi:10.1182/blood-2015-02-626291.
- Stanczak MA, Siddiqui SS, Trefny MP, Thommen DS, Boligan KF, von GS, Tzankov A, Tietze L, Lardinois D, Heinzelmann-Schwarz V, et al. Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J Clin Invest. 2018;128:4912–23. doi:10.1172/JCI120612.
- Haas Q, Boligan KF, Jandus C, Schneider C, Simillion C, Stanczak MA, Haubitz M, Jafari SMS, Zippelius A, Baerlocher GM, et al. Siglec-9 Regulates an Effector Memory CD8+ T-cell Subset That Congregates in the Melanoma Tumor Microenvironment. Cancer Immunol Res. 2019;7:707–18. doi:10.1158/2326-6066.CIR-18-0505.
- Wang J, Sun J, Liu LN, Flies DB, Nie X, Toki M, Zhang J, Song C, Zarr M, Zhou X, et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat Med. 2019;25:656–66. doi:10.1038/s41591-019-0374-x.
- Limagne E, Richard C, Thibaudin M, Fumet J-D, Truntzer C, Lagrange A, Favier L, Coudert B, Ghiringhelli F. Tim-3/galectin-9 pathway and mMDSC control primary and secondary resistances to PD-1 blockade in lung cancer patients. Oncoimmunology. 2019;8:e1564505. doi:10.1080/2162402X.2018.1564505.
- Gonçalves Silva I, Yasinska IM, Sakhnevych SS, Fiedler W, Wellbrock J, Bardelli M, Varani L, Hussain R, Siligardi G, Ceccone G, et al. The Tim-3-galectin-9 Secretory Pathway is Involved in the Immune Escape of Human Acute Myeloid Leukemia Cells. EBioMedicine. 2017;22:44–57. doi:10.1016/j.ebiom.2017.07.018.
- Wdowiak K, Gallego-Colon E, Francuz T, Czajka-Francuz P, Ruiz-Agamez N, Kubeczko M, Grochoła I, Wybraniec MT, Chudek J, Wojnar J. Increased serum levels of Galectin-9 in patients with chronic lymphocytic leukemia. Oncol Lett. 2019;17:1019–29. doi:10.3892/ol.2018.9656.
- Zheng Y, Feng W, Wang Y-J, Sun Y, Shi G, Yu Q. Galectins as potential emerging key targets in different types of leukemia. Eur J Pharmacol. 2019;844:73–78. doi:10.1016/j.ejphar.2018.11.019.
- Zhu J, Zheng Y, Zhang H, Liu Y, Sun H, Zhang P. Galectin-1 induces metastasis and epithelial-mesenchymal transition (EMT) in human ovarian cancer cells via activation of the MAPK JNK/p38 signalling pathway. Am J Transl Res. 2019;11:3862–78.
- Goud NS, Soukya PSL, Ghouse M, Komal D, Alvala R, Alvala M. Human Galectin-1 and its inhibitors: privileged target for cancer and HIV. Mini Rev Med Chem. 2019;19:1369–78. doi:10.2174/1389557519666190304120821.
- Chen H-Y, Fermin A, Vardhana S, Weng I-C, Lo KFR, Chang E-Y, Maverakis E, Yang R-Y, Hsu DK, Dustin ML, et al. Galectin-3 negatively regulates TCR-mediated CD4+ T-cell activation at the immunological synapse. Proc Natl Acad Sci U S A. 2009;106:14496–501. doi:10.1073/pnas.0903497106.
- Kaur M, Kumar D, Butty V, Singh S, Esteban A, Fink GR, Ploegh HL, Sehrawat S. Galectin-3 Regulates γ-Herpesvirus Specific CD8 T Cell Immunity. iScience. 2018;9:101–19. doi:10.1016/j.isci.2018.10.013.
- Polonskaya Z, Deng S, Sarkar A, Kain L, Comellas-Aragones M, McKay CS, Kaczanowska K, Holt M, McBride R, Palomo V, et al. T cells control the generation of nanomolar-affinity anti-glycan antibodies. J Clin Invest. 2017;127:1491–504. doi:10.1172/JCI91192.
- Li RE, van Vliet SJ, van Kooyk Y. Using the glycan toolbox for pathogenic interventions and glycan immunotherapy. Curr Opin Biotechnol. 2018;51:24–31. doi:10.1016/j.copbio.2017.11.003.
- Danishefsky SJ, Shue Y-K, Chang MN, Wong C-H. Development of Globo-H cancer vaccine. Acc Chem Res. 2015;48:643–52. doi:10.1021/ar5004187.
- Trial of Active Immunotherapy With OBI-833 (Globo H-CRM197) in Advanced/Metastatic Gastric, Lung, Colorectal or Breast Cancer Subjects - Tabular View - ClinicalTrials.gov; https://clinicaltrials.gov/ct2/show/record/NCT02310464
- Steentoft C, Migliorini D, King TR, Mandel U, June CH, Posey AD. Glycan-directed CAR-T cells. Glycobiology. 2018;28:656–69. doi:10.1093/glycob/cwy008.
- Mereiter S, Balmaña M, Campos D, Gomes J, Reis CA. Glycosylation in the Era of Cancer-Targeted Therapy: where Are We Heading?. Cancer Cell. 2019;36:6–16. doi:10.1016/j.ccell.2019.06.006.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. doi:10.1038/nrc3239.
- Van Cutsem E, Köhne C-H, Hitre E, Zaluski J, Chang Chien C-R, Makhson A, D’Haens G, Pintér T, Lim R, Bodoky G, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–17. doi:10.1056/NEJMoa0805019.
- Lee HT, Lee SH, Heo Y-S. Molecular Interactions of Antibody Drugs Targeting PD-1, PD-L1, and CTLA-4 in Immuno-Oncology. Molecules. 2019;24:1190.
- Munkley J, Scott E. Targeting Aberrant Sialylation to Treat Cancer. Medicines. 2019;6:102. doi:10.3390/medicines6040102.
- Fraschilla I, Pillai S. Viewing Siglecs through the lens of tumor immunology. Immunol Rev. 2017;276:178–91. doi:10.1111/imr.2017.276.issue-1.
- A safety and tolerability study of NC318 in subjects with advanced or metastatic solid tumors - Full Text View - clinicalTrials.gov [Internet]; [accessed 2019 Sep 18]. https://clinicaltrials.gov/ct2/show/NCT03665285
- Ren X. Immunosuppressive checkpoint Siglec-15: a vital new piece of the cancer immunotherapy jigsaw puzzle. Cancer Biol Med. 2019;16:205–10. doi:10.20892/j.issn.2095-3941.2018.0141.
- Natoni A, Macauley MS, O’Dwyer ME. Targeting Selectins and Their Ligands in Cancer. Front Oncol. 2016;6:93. doi:10.3389/fonc.2016.00093.
- Chou F-C, Chen H-Y, Kuo -C-C, Sytwu H-K. Role of Galectins in Tumors and in Clinical Immunotherapy. Int J Mol Sci. 2018;19. doi:10.3390/ijms19020430.
- He Y, Cao J, Zhao C, Li X, Zhou C, Hirsch FR. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther. 2018;11:7005–09. doi:10.2147/OTT.
- Van Landuyt L, Lonigro C, Meuris L, Callewaert N. Customized protein glycosylation to improve biopharmaceutical function and targeting. Curr Opin Biotechnol. 2019;60:17–28. doi:10.1016/j.copbio.2018.11.017.
- Buettner MJ, Shah SR, Saeui CT, Ariss R, Yarema KJ. Improving Immunotherapy Through Glycodesign. Front Immunol. 2018;9:2485.
- Nemudryi AA, Valetdinova KR, Medvedev SP, Zakian SM. TALEN and CRISPR/Cas Genome Editing Systems: tools of Discovery. Acta Naturae. 2014;6:19–40. doi:10.32607/20758251-2014-6-3-19-40.
- Gupta RM, Musunuru K. Expanding the genetic editing tool kit: zFNs, TALENs, and CRISPR-Cas9. J Clin Invest. 2014;124:4154–61. doi:10.1172/JCI72992.
- Komor AC, Badran AH, Liu DR. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell. 2017;169:559. doi:10.1016/j.cell.2017.04.005.
- Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–57. doi:10.1038/s41586-019-1711-4.
- Kuball J, Hauptrock B, Malina V, Antunes E, Voss R-H, Wolfl M, Strong R, Theobald M, Greenberg PD. Increasing functional avidity of TCR-redirected T cells by removing defined N-glycosylation sites in the TCR constant domain. J Exp Med. 2009;206:463–75. doi:10.1084/jem.20082487.
- Okada M, Chikuma S, Kondo T, Hibino S, Machiyama H, Yokosuka T, Nakano M, Yoshimura A. Blockage of Core Fucosylation Reduces Cell-Surface Expression of PD-1 and Promotes Anti-tumor Immune Responses of T Cells. Cell Rep. 2017;20:1017–28. doi:10.1016/j.celrep.2017.07.027.
- Cedeno-Laurent F, Opperman M, Barthel SR, Hays D, Schatton T, Zhan Q, He X, Matta KL, Supko JG, Frank MH, et al. Metabolic inhibition of galectin-1-binding carbohydrates accentuates anti-tumor immunity. J Invest Dermatol. 2012;132:410–20. doi:10.1038/jid.2011.335.
- Ito K, Scott SA, Cutler S, Dong L-F, Neuzil J, Blanchard H, Ralph SJ. Thiodigalactoside inhibits murine cancers by concurrently blocking effects of galectin-1 on immune dysregulation, angiogenesis and protection against oxidative stress. Angiogenesis. 2011;14:293–307. doi:10.1007/s10456-011-9213-5.
- Rillahan CD, Antonopoulos A, Lefort CT, Sonon R, Azadi P, Ley K, Dell A, Haslam SM, Paulson JC. Global Metabolic Inhibitors of Sialyl- and Fucosyltransferases. Nat Chem Biol. 2012;8:661–68. doi:10.1038/nchembio.999.
- Büll C, Boltje TJ, Balneger N, Weischer SM, Wassink M, Gemst JJV, Bloemendal VRLJ, Boon L, Vlag JVD, Heise T, et al. Sialic Acid Blockade Suppresses Tumor Growth by Enhancing T-cell-Mediated Tumor Immunity. Cancer Res. 2018;78:3574–88. doi:10.1158/0008-5472.CAN-17-3376.
- Dias AM, Correia A, Pereira MS, Almeida CR, Alves I, Pinto V, Catarino TA, Mendes N, Leander M, Oliva-Teles MT, et al. Metabolic control of T cell immune response through glycans in inflammatory bowel disease. Proc Natl Acad Sci. 2018; 115:E4651–60. doi:10.1073/pnas.1720409115.
- Zheng T, Jiang H, Gros M, Del Amo DS, Sundaram S, Lauvau G, Marlow F, Liu Y, Stanley P, Wu P. Tracking N-acetyllactosamine on cell-surface glycans in vivo. Angew Chem Int Ed Engl. 2011;50:4113–18. doi:10.1002/anie.v50.18.
- Li J, Chen M, Liu Z, Zhang L, Felding BH, Moremen KW, Lauvau G, Abadier M, Ley K, Wu P. A Single-Step Chemoenzymatic Reaction for the Construction of Antibody-Cell Conjugates. ACS Cent Sci. 2018;4:1633–41. doi:10.1021/acscentsci.8b00552.
- Parmar S, Liu X, Najjar A, Shah N, Yang H, Yvon E, Rezvani K, McNiece I, Zweidler-McKay P, Miller L, et al. Ex vivo fucosylation of third-party human regulatory T cells enhances anti-graft-versus-host disease potency in vivo. Blood. 2015;125:1502–06. doi:10.1182/blood-2014-10-603449.
- Cabral J, Hanley SA, Gerlach JQ, O’Leary N, Cunningham S, Ritter T, Ceredig R, Joshi L, Griffin MD. Distinctive Surface Glycosylation Patterns Associated With Mouse and Human CD4+ Regulatory T Cells and Their Suppressive Function. Front Immunol. 2017;8:987. doi:10.3389/fimmu.2017.00987.
- Sadighi Akha AA, Berger SB, Miller RA. Enhancement of CD8 T-cell function through modifying surface glycoproteins in young and old mice. Immunology. 2006;119:187–94. doi:10.1111/imm.2006.119.issue-2.
- Yoon HY, Koo H, Kim K, Kwon IC. Molecular imaging based on metabolic glycoengineering and bioorthogonal click chemistry. Biomaterials. 2017;132:28–36. doi:10.1016/j.biomaterials.2017.04.003.
- Büll C, Boltje TJ, Wassink M, de Graaf AMA, van Delft FL, den Brok MH, Adema GJ. Targeting aberrant sialylation in cancer cells using a fluorinated sialic acid analog impairs adhesion, migration, and in vivo tumor growth. Mol Cancer Ther. 2013;12:1935–46. doi:10.1158/1535-7163.MCT-13-0279.
- Varki A, Gagneux P. Multifarious roles of sialic acids in immunity. Ann N Y Acad Sci. 2012;1253:16–36. doi:10.1111/j.1749-6632.2012.06517.x.
- Büll C, Heise T, van Hilten N, Pijnenborg JFA, Bloemendal VRLJ, Gerrits L, Kers-Rebel ED, Ritschel T, den Brok MH, Adema GJ, et al. Steering Siglec-Sialic Acid Interactions on Living Cells using Bioorthogonal Chemistry. Angew Chem Int Ed Engl. 2017;56:3309–13. doi:10.1002/anie.201612193.
- Pan H, Li P, Li G, Li W, Hu B, He H, Chen Z, Wang F, Liu L, Gong Y, et al. Glycometabolic Bioorthogonal Chemistry‐Guided Viral Transduction for Robust Human T Cell Engineering. Adv Funct Mater. 2019;29:1807528. doi:10.1002/adfm.v29.22.
- Hong S, Shi Y, Wu NC, Grande G, Douthit L, Wang H, Zhou W, Sharpless KB, Wilson IA, Xie J, et al. Bacterial glycosyltransferase-mediated cell-surface chemoenzymatic glycan modification. Nat Commun. 2019;10:1–11. doi:10.1038/s41467-019-09608-w.
- Mondal N, Silva M, Castano AP, Maus MV, Sackstein R. Glycoengineering of chimeric antigen receptor (CAR) T-cells to enforce E-selectin binding. J Biol Chem. 2019;294:18465–74. doi:10.1074/jbc.RA119.011134.
- Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–30. doi:10.1038/nature21349.