
ABSTRACT
Over the last decades, the use of phylogenetic methods in the study of emerging infectious diseases has gained considerable traction in public health. Particularly, the integration of phylogenetic analyses with the understanding of the pathogen dynamics at the population level has provided powerful tools for epidemiological surveillance systems. In the same way, the development of statistical methods and theory, as well as improvement of computational efficiency for evolutionary analysis, has expanded the use of these tools for vaccine and antiviral development. Today with the Coronavirus Disease 2019 (COVID-19), this seems to be critical. In this article, we discuss how the application of phylodynamic analysis can improve the understanding of current pandemic dynamics as well as the design, selection, and evaluation of vaccine candidates and antivirals.
Introduction
Recent developments in phylogenetic methods have made it possible to detect and track evolutionary changes in pathogens in time and space, constituting an evolving branch of phylogenetics called phylodynamics.Citation1 The development and application of coalescent theory in this field have allowed the integration of phylogenetic analysis with traditional epidemiological analysis in the understanding of the geospatial and temporal pathogen dynamics at the population level.Citation1,Citation2 This integration allows one to infer geospatial transmission patterns and networks, as well as epidemiological parameters of infectivity, and spread of disease when used at the “between host” level. Additionally, the construction of a viral demographic model based on viral diversity and its coalescence could be used also in assessing control measures like vaccines or therapeutics.Citation3
The coalescent theory is based on a mathematical model for lineage separation and genetic drift that allows one to analyze genetic variation as a stochastic process, describing population events retrospectively.Citation4 In viral evolution, this theory permits analysis of genetic diversity by testing several models based on evolutionary processes (genetic drift, mutation, natural selection, etc.) and makes inferences about changes in viral population size, immune selection, and spatial dynamics.Citation5–7 Phylodynamics applies the coalescent theory to describe the relationship between a population’s genetic history and the shared ancestry of randomly sampled individuals and uses the molecular clock concept to infer the timing of intersecting phylogenic events.Citation8
Therefore, phylodynamics details certain aspects of viral demography like rates of viral population growth and decline, the distribution of branching events in phylogenetic trees, population structure, and immune selection. It also enables the estimation of the basic reproductive number (R0) directly from gene sequence data, providing a link between evolutionary analysis of genome sequences and the epidemiology of an infectious disease.Citation2 In the context of the current pandemic, the analysis of the evolutionary relationship of genes or genomes with spatiotemporal variables has allowed identifying the possible origin of the cross-species transmission of the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), causing the Coronavirus Disease 2019 (COVID-19), and the geographical patterns of infection migration.Citation9
On the other hand, patterns inferred from phylodynamic analysis allow making inferences about the immune response of the host. When the viral diversity is analyzed within an individual (within host level) and at the population level (between host level), and contrasted with the level of conferred immunity, one can infer the level of cross-immunity between different genotypes and the level of immune selective pressure.Citation10 In the context of the current SARS-CoV-2 pandemic, these data could be used for making inferences and predictions about the behavior of this emerging infection and the effectiveness of vaccination strategies in the short, mid, and long term, as has been used for influenza.Citation11,Citation12 Moreover, the antiviral design could be improved and the effectiveness of viral control measures over time could be assessed.Citation8
Although historically the study of the phylodynamic of pathogens has been limited by the cost and timescale of the generation of molecular data, during this pandemic, genomic data about SARS-CoV-2 have been widely and freely available ().Citation15,Citation16 These, along with the possibility of performing next-generation sequencing (NGS) to reveal the genetic variation present among viruses in a single sample, can provide information about the viral diversity at the individual level for vaccine and therapeutics design. In this article, we discuss how the application of phylodynamic analysis has helped not only in the understanding of current pandemic dynamics but also how it could help in the design, selection, and evaluation of vaccine candidates and antivirals.
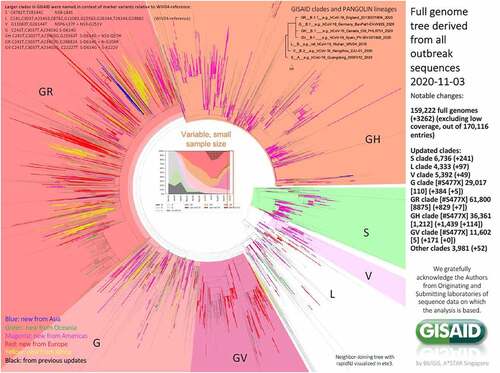
Figure 1. Phylogenetic clades classification of SARS-CoV-2 based on full-genome sequences by GISAID.Citation13,Citation14
Using phylodynamics for infectious disease modeling and understanding the SARS-CoV-2 pandemic
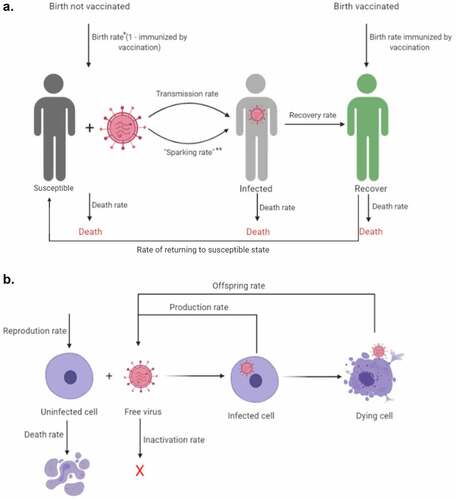
Under the family of SIR models, populations are usually divided into at least three compartments.Citation17 These three compartments are 1. Susceptible (S) that corresponds to nonimmune non-exposed hosts that can get infected, 2. Infected (I) that corresponds to individuals infected that can spread the disease, and 3. Recovered (R) that corresponds to individuals that recover or are “removed” from the population, this is, they get longstanding immunity for a timeframe or die. Some models under this family, add compartments, like the E compartment, under the SEIR model, that corresponds to Exposed individuals that do not spread the infection, since they have a latent asymptomatic infection but are not infectious, which is not the case for SARS-CoV-2 given that asymptomatic patients can also spread the disease (), and also, can consider the rate of becoming susceptible again after infection, as would be important in the case of reinfection (), a plausible but uncertain phenomenon in the case of SARS-CoV-2,Citation17 and the rate of infection from different human to human sources, like contaminated surfaces (sparking rate, ), as has been documented for SARS-CoV-2 and other viruses.Citation18
Figure 2. Schematic diagram of between-host (a) and intra-host virus (b) dynamics. Arrows correspond to different rates, while pictures correspond to different states of the viral SIR model created with BioRender.com. *In this case, birth rate corresponds to the rate at which new individuals born, **Sparking rate corresponds to transmission from outside non-human to human sources, as it has been documented for SARS-CoV-2 and other viruses
Under these models, phylodynamic inference using Bayesian statistics allows us to estimate the size of each compartment based on the analysis of viral diversity and its coalescence back in time.Citation19 Under the assumption of strict or relaxed molecular clock models, different approaches could be implemented for estimation of the number of susceptible, infected, and recovered cases using open resources like BEAST or R.Citation20–23 These kinds of analysis require as input nucleotide sequences alignment of genic or whole-genome data sampled serially over the time or sampled at one time point. For each sequence, sampling date is specified, and this information with a scheme of substitutions and a molecular clock model helps us to estimate the timescale of the tree and the rate of evolution as it connects the temporal information contained in the sampling times to the genetic similarities embedded in the sequences.Citation24 Additional to these parameters, we also choose a distribution model that describes how the population size is expected to change over time.
This method provides estimations of the infection rate (births), the recovery rate (deaths), and the population size (N).Citation20 Under the birth–death SIR model, a constant birth-death rate is used to model the spread of an epidemic and to model speciation and extinction. Under the epidemiology view, a birth event is a transmission and a death event is a recovery or death.Citation19 The information provided by the branching times and the molecular clock rate can be used for estimation of the time to the most recent ancestor (TMRA), which provides information regarding the date of introduction of the virus to a specific geographic area.Citation22
The outputs of this analysis are used for providing important estimations about epidemiological parameters like the basic reproductive number (R0) which corresponds to the total number of secondary cases caused by an infectious individual when introduced into a fully susceptible population, the critical community size (CCS) which is the minimum number of individuals in a population required for a pathogen to persist in a population, and the critical vaccination threshold (pc), which corresponds to the proportion of the population that must have immunity to make the epidemic disappear.Citation2,Citation17 Since the control of an epidemic relies on the reduction of the R0 to <1, the pc can be calculated based on the calculation of the R0. and the estimates of the real burden of the disease could also be assessed through the estimation of the epidemic size.Citation25,Citation26
In the context of the current pandemic, phylodynamic analysis has not only analyzed and estimated those parameters but also has been in good agreement with epidemiological observations.Citation9,Citation27 The origin of the outbreak has been tracked by epidemiological and phylodynamic analyses to Wuhan, China, between November and December 2019 (95% CI: November 21-December 20, 2019).Citation9,Citation27 In agreement with epidemiological data, the estimated R0 has been 2.15 (95%CI: 1.79–2.75), with a doubling time of 7.1 days (95% CI: 3.0–20.5).Citation28–30 In Latin America for instance, the contrast of epidemiologically observed and phylodynamically inferred R0 has shown agreement and has been used for temporal analysis of the introduction of SARS-CoV-2 suggesting a detection lag of at least 21 days in some countries, supporting the complementary approaches of epidemiological and genomic surveillance.Citation31 In this sense, the COVID-19 pandemic and the rapid availability of genomic information of the SARS-CoV-2 virus have represented a unique opportunity for contrasting observations.
In this regard, other studies have shown how phylodynamic inference can be used in tracking transmission chainsCitation32–34 and evaluating the effectiveness of public health measures through the assessment of the evolution of R0 in time.Citation32,Citation33 Evaluation of transmission chains in Australia through cluster analysis showed good agreement between epidemiological and genomic clusters,Citation32 and phylodynamic analysis at the country level in Brazil has shown the role of large and highly connected populated cities in the establishment of SARS-CoV-2.Citation33 Therefore, phylodynamic analysis can be used as a complement to traditional surveillance methods. Additionally, this analysis can serve as a tool for evaluating transmission chains and public health measures for disease control.
Use of phylodynamics in vaccine design for preventing SARS-CoV-2 infection
Viral virulence, diversity, and fitness represent a challenge for viral vaccine development.Citation3 These are the most important factors in viral evolution, and phylogenetic analysis is a key tool to simultaneously evaluate these factors.Citation35 Diversity of RNA viruses can appear over a short time since they are prone to replication errors, generating heterogeneous viral populations.Citation3 Moreover, antigenic drift and shift, and the bottleneck at the moment of transmission, have an impact on their evolutionary dynamics and determine therapeutic effectiveness, resistance to antivirals, and evasion of the immune response.Citation36
However, compared to other RNA viruses, coronaviruses exhibit relatively higher replication fidelity. For example, the SARS-CoV mutation rate was estimated at 9.0 × 10−7 substitutions per nucleotide per replication cycle,Citation37,Citation38 while most RNA viruses have a rate of 1 × 10−3 to 1 × 10−5 substitutions per nucleotide per replication cycle.Citation39 Therefore, it seems that SARS-CoV-2 is not mutating significantly as it spreads,Citation40 which could have important implications for vaccine design.
Although evidence for antigenic drift or shift for SARS-CoV-2 is limited, the sustained human-to-human transmission could permit the acquisition of mutations with fitness advantages and immunological resistance.Citation36 Analysis of SARS-CoV-2 genomics data have shown evidence of human clonal evolution. In this sense, SARS-CoV-2 genomes analysis has shown mutations that have emerged independently in multiple times.Citation41 Interestingly, three sites in Orf1ab and one in Spike protein have experienced numerous recurrent mutations (>15 events) that are of particular interest in the context of adaptation of SARS-CoV-2 to the human host.Citation41 Consistent with that, characterizing frequently mutated residues by aligning ~660 SARS-CoV-2 genomes and validated in 10,000 datasets has shown that Orf1ab and Spike protein contained numerous nonsynonymous mutations, after available in GISAID Nextstrain.Citation42 However, the degree to which these observed changes are related to antigenic escape or if SARS-CoV-2 may undergo antigenic drift is unclear and has not been documented beyond in vitro experiments,Citation43 and inferred by the analysis of variants and glycosylation sites.Citation44,Citation45
Moreover, once a vaccine for SARS-CoV-2 is developed, it will be important to continue monitoring the genetic diversity and population dynamics of circulating strains to assess their adaptive evolution. At this point, phylodynamics provides a powerful approach for updating vaccines by analyzing evolutionary dynamics and predicting evolutionary changes in populations. Similar approaches have used with the influenza vaccine, in which the assessment of the distribution of variants and its departure from neutrality has helped in predicting emerging lineages and escape variants,Citation12,Citation46 becoming an integral part of the yearly vaccine design cycle.Citation35 As another example, phylodynamic approaches have been used as a continuous surveillance tool to evaluate the efficacy of the vaccine for group A rotaviruses, assessing the genotypic diversity and mutation of antigenic epitopes, compared to the prevalent genotypes of the vaccine strains.Citation47
Currently, different approaches such as recombinant proteins, viral vectors, DNA, RNA, and live attenuated vaccines (LAV) are being developed as SARS-CoV-2 vaccine candidates, with at least 172 candidates in pre-clinical evaluation, 61 in clinical trials, and 2 vaccines authorized for emergency use by the Food and Drug Administration in the USA (Pfizer-BioNTech COVID-19 Vaccine and Moderna COVID-19 Vaccine).Citation48 A critical goal of vaccine design is to elicit a similar immune response compared with the natural infection, but also to produce a wide neutralizing immune response against different genotypes to produce a durable and protective response. In the case of vaccines against SARS-CoV-2, the strategies aim to induce neutralizing antibodies, which are directed to the viral spike (S) protein as it mediates virus attachment and entry to host cell, and mainly to the RBD domain of this protein.Citation49,Citation50
Single nucleotide polymorphism (SNP) genotyping of SARS-CoV-2 has revealed that some of the more common SNP mutations are located in the S protein, like the 23403A > G spike glycoprotein mutation D614G, but their implications in pathogenesis and immune escape are not clear.Citation51 While this variant has been effectively neutralized by isolated monoclonal neutralizing antibodies, other variants with mutations in the receptor-binding domain (RBD) showed complete resistance to one of the tested antibodies, highlighting the impact of viral genetic diversity on the ability to produce a broadly neutralizing response.Citation52 However, little is known about the features of the immune response during natural infection with SARS-CoV-2. The kinetics of neutralizing antibodies suggest a decline after three months, and there are reports consistent with the possibility of reinfection.Citation53–57
In this point, the analysis of phylodynamic patterns can be used to infer the level of cross-immunity among different viral clades or lineages, since phylogenies of the virus will be structured depending on the level of cross-immunity between different genotypes or variants. Grenfell et al. have previously discussed the role of immunity in structuring phylodynamic patterns.Citation10 Briefly, viruses that induce a high cross-immunity across different genotypes will have a phylogeny with a strong spatial structure. In contrast, viruses that induce partial cross-immunity across genotypes will have a phylogeny structured through time, and viruses that produce immune enhancement will have chaotic or complex dynamics.Citation10 Therefore, although there is no evidence of lack of cross-immunity among SARS-CoV-2, analyzing the structure of the obtained phylogeny would provide insights in this regard between genetic variants. At the same time, information can be gathered from the level of immune response and the number of viral copies through time in groups of individuals. This allows elucidating the Evolutionary Immunity Profile (EIP), which provides information about the probability for a host to be a major source of host-selected variants.Citation10 The analysis of genome regions and their rate of evolution would allow to identify regions under selective pressure and to suggest new antigenic determinants targeted by neutralizing antibodies.
In this context, a phylodynamic approach could be used to test if the immune response is operating as a selection pressure and diversity accumulated in this region driven by immune escape. The effect of directional selection driven by immune escape can be inferred from the phylogenetic tree balance. For example, in a phylodynamic analysis, the ladder-like shape of a viral phylogeny reveals the hallmarks of selection driven by immune escape. In contrast, a more balanced phylogeny may occur when a virus is not subject to immune selection or other sources of directional selection,Citation8 although this inference should be taken with caution because these phylogenetic patterns could also reflect sequential bottlenecks occurring with a rapid spatial spread of some types of virus. However, phylogenetic supported with statistical analysis could confirm viral lineages with a larger number of positively selected sites and predicting evolutionary changes, which are important for vaccine re-design.Citation3
Taken together, the information obtained through phylodynamic analysis can enhance vaccine design by 1. Predicting the targets of immune response for neutralizing infection, 2. Predicting the level of cross-immunity produced during natural infection, 3. Evaluating the role of vaccine-induced immunity as a selective pressure.
Use of phylodynamic in antiviral research for treating SARS-CoV-2 infection
Just as an epidemic/pandemic is understood through an SIR model, viral infection within a host can be understood in the same way. The viral infection is treated as a microepidemic among host cells. Those cells are treated as susceptible (S) individuals that get infected (I) and then are removed (R) as they die ().Citation3 Whether the virus infection will spread within the host depends on a condition very similar to the spread of an epidemic at the population level and would depend on the rate at which susceptible cells are infected, and the rate at which infected cells die or are removed by the immune response or the cytotoxic effect of the virus.Citation3 To propagate the infection in the host, the conditions of viral dynamics must favor R0 > 1, and hence therapeutic interventions such as antivirals should seek to reduce R0 below 1 to promote infection control by reducing the rate at which cells get infected.Citation58 At the same time, RNA viruses can rapidly adapt to changes in the host environment, since they exist as genetically diverse quasispecies.Citation59 SARS-CoV-2 is not the exemption, and sequence analysis indicated the presence of viral quasispecies, as also reported for SARS-CoV and MERS-CoV.Citation60
The assessment of viral diversity in antiviral experiments, both in vitro and in vivo, can help to estimate their effect on R0 through the analysis of their effect on diversity. This diversity could be also tested looking for evidence of positive selection and directional selection, through the use of probabilistic models of accumulating mutations, assessing the genetic barrier, but also analyzing the diversity control that must be achieved to maintain an R0 < 1. Depending on the antiviral effect on diversity, the need of using multiple drugs and regimens for disease control could be analyzed, and also the probability of escape variants based on the understanding of viral divergence and the substitution rate could be determined.Citation3,Citation61,Citation62
Different drugs have been tested against SARS-CoV-2 as antivirals, both as therapeutics and prophylactics including biological therapy, immune-therapeutics, and antiretrovirals.Citation63–68 The absence of effective antiviral drugs for the treatment of SARS-CoV-2 infection has led to the evaluation of drugs already FDA-approved for the treatment of other viral infections such as lopinavir, ritonavir, ribavirin, remdesivir,Citation69 oseltamivir, favipiravir, umifenovir,Citation67 penciclovir, and chloroquine.Citation70,Citation71 When the introduction of an approved drug happens, phylodynamic analysis can provide tools for monitoring the effectiveness of the intervention by analyzing and quantifying how intra-host viral evolution over time in serial samples and the effect of cessation of replication with effective treatment.Citation8
Therefore, the use of the coalescent model would allow the estimation of the effective population size (Ne), which in terms of phylodynamic is proportional to the effective number of infected population,Citation72 and the assessment of this parameter would help to identify the effect of the introduction of antiviral therapy in the epidemic curve and transmission dynamics.Citation73 Additionally, although antiviral resistance is more unlikely for an acute virus infection as COVID-19 than for a chronic infection like HIV, the example of anti-retroviral therapy (ART) has shown that evolutionary analysis can be used for the detection of adaptive mutations that if fixed can promote antiviral resistance.Citation74 This approach could also be used for timely detection of the emergence of drug-resistant strains and the need to switch to a new three-drug regimen (switching therapy).Citation73 Coupling these data with clinical and epidemiological metadata would help also to identify transmission networks associated with the emergence of resistance in case it happens.Citation74
Conclusions
The development of computational and statistical techniques and their application for the understanding of the relationship between spatiotemporal variables (i.e., geographical area, dates) and the genomic diversity of pathogens like SARS-CoV-2 can provide important insights in the understanding of the current pandemics, but also in the design and evaluation of interventions for prevention and treatment of infection. The use of these methods has been historically constrained by the availability of genomic data of the pathogens. However, initiatives like EpiCoVTM by GISAID have made genomic information about the virus free and widely available.Citation13,Citation75 Given the implications of being able to design, assess, and adapt vaccines through the use of genomic data and computational diversity analysis, local authorities should invest in sequencing technologies for genomic surveillance of current pandemics,Citation76 especially in developing countries where this technology is not widely available. As the pandemic has expanded, studies contrasting traditional epidemiological surveillance and phylodynamics have emerged. However, this review has been limited by the amount of evidence linking phylodynamic analysis with host immune response and the lack of studies using this approach for the assessment of antiviral drugs. Given the potential benefits of these analyses in the design of vaccines and antivirals, further research is recommended.
Acknowledgments
We thank Dr. Matthew H. Collins at Hope Clinic of the Emory Vaccine Center, Division of Infectious Diseases, School of Medicine, Emory University for English revision.
Disclosure statement
The authors declare no competing interests.
References
- Rife BD, Mavian C, Chen X, Ciccozzi M, Salemi M, Min J, Prosperi MC. Phylodynamic applications in 21(st) century global infectious disease research. Glob Health Res Policy. 2017;2:13. doi:10.1186/s41256-017-0034-y.
- Holmes EC. Evolutionary history and phylogeography of human viruses. Annu Rev Microbiol. 2008;62:307–28. doi:10.1146/annurev.micro.62.081307.162912.
- Ojosnegros S, Beerenwinkel N. Models of RNA virus evolution and their roles in vaccine design. Immunome Res. 2010;6(Suppl 2):S5. doi:10.1186/1745-7580-6-S2-S5.
- Templeton AR. Genetic drift in large populations and coalescence. Populat Gen Microevolution Theory. 2006;118–67.
- Biswas NK, Majumder PP. Analysis of RNA sequences of 3636 SARS-CoV-2 collected from 55 countries reveals selective sweep of one virus type. Indian J Med Res. 2020;151:450–58. doi:10.4103/ijmr.IJMR_1125_20.
- Nie Q, Li X, Chen W, Liu D, Chen Y, Li H, Li D, Tian M, Tan W, Zai J, et al. Phylogenetic and phylodynamic analyses of SARS-CoV-2. Virus Res. 2020;287:198098. doi:10.1016/j.virusres.2020.198098.
- Sironi M, Hasnain SE, Rosenthal B, Phan T, Luciani F, Shaw MA, Sallum MA, Mirhashemi ME, Morand S, González-Candelas F, et al. SARS-CoV-2 and COVID-19: a genetic, epidemiological, and evolutionary perspective. Infect Genet Evol. 2020;84:104384. doi:10.1016/j.meegid.2020.104384.
- Volz EM, Koelle K, Bedford T. Viral phylodynamics. PLoS Comput Biol. 2013;9:e1002947. doi:10.1371/journal.pcbi.1002947.
- Benvenuto D, Giovanetti M, Salemi M, Prosperi M, De Flora C, Junior Alcantara LC, Angeletti S, Ciccozzi M. The global spread of 2019-nCoV: a molecular evolutionary analysis. Pathog Glob Health. 2020;114:64–67. doi:10.1080/20477724.2020.1725339.
- Grenfell BT, Pybus OG, Gog JR, Wood JL, Daly JM, Mumford JA, Holmes EC. Unifying the epidemiological and evolutionary dynamics of pathogens. Sci. 2004;303:327–32. doi:10.1126/science.1090727.
- Lei N, Wang HB, Zhang YS, Zhao JH, Zhong Y, Wang YJ, Huang L-Y, Ma J-X, Sun Q, Yang L, et al. Molecular evolution of influenza B virus during 2011-2017 in Chaoyang, Beijing, suggesting the free influenza vaccine policy. Sci Rep. 2019;9:2432. doi:10.1038/s41598-018-38105-1.
- Bush RM, Bender CA, Subbarao K, Cox NJ, Fitch WM. Predicting the evolution of human influenza A. Science. 1999;286:1921–25. doi:10.1126/science.286.5446.1921.
- GISAID. Genomic epidemiology of novel coronavirus; 2020. [accessed 2020 Nov 05]. https://nextstrain.org/ncov
- Shu Y, McCauley J. GISAID: global initiative on sharing all influenza data - from vision to reality. Euro Surveill. 2017;22(13):30494.
- Elbe S, Buckland-Merrett G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob Chall. 2017;1:33–46. doi:10.1002/gch2.1018.
- Rodriguez-Morales AJ, Balbin-Ramon GJ, Rabaan AA, Sah R, Dhama K, Paniz-Mondolfi A, Pagliano P, Esposito S. Genomic epidemiology and its importance in the study of the COVID-19 pandemic. Infez Med. 2020;28:139–42.
- Evans AS. Viral infections of humans: epidemiology and control. Springer Sci Business Media. 2013.
- Azuma K, Yanagi U, Kagi N, Kim H, Ogata M, Hayashi M. Environmental factors involved in SARS-CoV-2 transmission: effect and role of indoor environmental quality in the strategy for COVID-19 infection control. Environ Health Prev Med. 2020;25:66. doi:10.1186/s12199-020-00904-2.
- Kuhnert D, Stadler T, Vaughan TG, Drummond AJ. Simultaneous reconstruction of evolutionary history and epidemiological dynamics from viral sequences with the birth-death SIR model. J R Soc Interface. 2014;11:20131106. doi:10.1098/rsif.2013.1106.
- Volz EM, Siveroni I. Bayesian phylodynamic inference with complex models. PLoS Comput Biol. 2018;14:e1006546. doi:10.1371/journal.pcbi.1006546.
- Volz EM, Romero-Severson E, Leitner T. Phylodynamic Inference across Epidemic Scales. Mol Biol Evol. 2017;34:1276–88. doi:10.1093/molbev/msx077.
- Volz E, Frost S. Scalable relaxed clock phylogenetic dating. Virus Evolution. 2017;3. doi:10.1093/ve/vex014.
- Bouckaert R, Vaughan TG, Barido-Sottani J, Duchêne S, Fourment M, Gavryushkina A, Heled J, Jones G, Kühnert D, De Maio N, et al. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2019;15(4):e1006650. doi:10.1371/journal.pcbi.1006650.
- Drummond AJ, Pybus OG, Rambaut A, Forsberg R, Rodrigo AG. Measurably evolving populations. Trends Ecol Evol. 2003;18:481–88. doi:10.1016/S0169-5347(03)00216-7.
- Magiorkinis G, Sypsa V, Magiorkinis E, Paraskevis D, Katsoulidou A, Belshaw R, Fraser C, Pybus OG, Hatzakis A, et al. Integrating phylodynamics and epidemiology to estimate transmission diversity in viral epidemics. PLoS Comput Biol. 2013;9:e1002876–e. doi:10.1371/journal.pcbi.1002876.
- Rasmussen DA, Boni MF, Koelle K. Reconciling phylodynamics with epidemiology: the case of dengue virus in southern Vietnam. Mol Biol Evol. 2014;31:258–71. doi:10.1093/molbev/mst203.
- Volz E, Baguelin M, Bhatia S, Boonyasiri A, Cori A, Cucunubá Z, Cuomo-Dannenburg G, Donnelly CA, Dorigatti I, FitzJohn R. Report 5: Phylogenetic analysis of SARS-CoV-2.
- Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, Ren R, Leung KSM, Lau EHY, Wong JY, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med. 2020;382:1199–207. doi:10.1056/NEJMoa2001316.
- Park M, Cook AR, Lim JT, Sun Y, Dickens BL. A systematic review of COVID-19 epidemiology based on current evidence. J Clin Med. 2020;9(4):967.
- Wu JT, Leung K, Leung GM. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: a modelling study. Lancet. 2020;395:689–97. doi:10.1016/S0140-6736(20)30260-9.
- Rojas-Gallardo DM, Garzon-Castano SC, Millan N, Jimenez-Posada EV, Martinez-Gutierrez M, Ruiz-Saenz J, Cardona-Ospina JA. COVID-19 in Latin America: contrasting phylodynamic inference with epidemiological surveillance. (Molecular epidemiology of COVID-19 in Latin America). medRxiv 2020; 2020.05.23.20111443.
- Seemann T, Lane CR, Sherry NL, Duchene S, Gonçalves da Silva A, Caly L, Sait M, Ballard SA, Horan K, Schultz MB, et al. Tracking the COVID-19 pandemic in Australia using genomics. Nat Commun. 2020;11:4376. doi:10.1038/s41467-020-18314-x.
- Candido DS, Claro IM, de Jesus JG, Souza WM, Moreira FRR, Dellicour S, Mellan TA, du Plessis L, Pereira RHM, Sales FCS, et al. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science. 2020; 369:1255–60.
- Lemey P, Hong S, Hill V, Baele G, Poletto C, Colizza V, et al. Accommodating individual travel history, global mobility, and unsampled diversity in phylogeography: a SARS-CoV-2 case study. bioRxiv. 2020.
- Ciccozzi M, Lai A, Zehender G, Borsetti A, Cella E, Ciotti M, Sagnelli E, Sagnelli C, Angeletti S. The phylogenetic approach for viral infectious disease evolution and epidemiology: an updating review. J Med Virol. 2019;91:1707–24. doi:10.1002/jmv.25526.
- Mandary MB, Masomian M, Poh CL. Impact of RNA virus evolution on Quasispecies formation and virulence. Int J Mol Sci. 2019;20. doi:10.3390/ijms20184657.
- Zhao Z, Li H, Wu X, Zhong Y, Zhang K, Zhang Y-P, Boerwinkle E, Fu Y-X. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol Biol. 2004;4:21. doi:10.1186/1471-2148-4-21.
- Eckerle LD, Becker MM, Halpin RA, Li K, Venter E, Lu X, Scherbakova S, Graham RL, Baric RS, Stockwell TB, et al. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010;6(5):e1000896. doi:10.1371/journal.ppat.1000896.
- Duffy S, Shackelton LA, Holmes EC. Rates of evolutionary change in viruses: patterns and determinants. Nat Rev Genet. 2008;9:267–76. doi:10.1038/nrg2323.
- Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, Hengartner N, Giorgi EE, Bhattacharya T, Foley B, et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182(4):812–27.e19. doi:10.1016/j.cell.2020.06.043.
- van Dorp L, Acman M, Richard D, Shaw LP, Ford CE, Ormond L, Owen CJ, Pang J, Tan CCS, Boshier FAT, et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect Genet Evol. 2020:104351. doi:10.1016/j.meegid.2020.104351.
- Laha S, Chakraborty J, Das S, Manna SK, Biswas S, Chatterjee R. Characterizations of SARS-CoV-2 mutational profile, spike protein stability and viral transmission. Infect Genet Evol. 2020;85:104445. doi:10.1016/j.meegid.2020.104445.
- Weisblum Y, Schmidt F, Zhang F, DaSilva J, Poston D, Lorenzi JC, Muecksch F, Rutkowska M, Hoffmann -H-H, Michailidis E, et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife. 2020;9:e61312. doi:10.7554/eLife.61312.
- Li Q, Wu J, Nie J, Zhang L, Hao H, Liu S, Zhao C, Zhang Q, Liu H, Nie L, et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell. 2020;182(5):1284–94 e9. doi:10.1016/j.cell.2020.07.012.
- Koyama T, Weeraratne D, Snowdon JL, Parida L. Emergence of drift variants that may affect COVID-19 vaccine development and antibody treatment. Pathogens. 2020;9:324. doi:10.3390/pathogens9050324.
- Rosenberg NA, Nordborg M. Genealogical trees, coalescent theory and the analysis of genetic polymorphisms. Nat Rev Genet. 2002;3:380–90. doi:10.1038/nrg795.
- Nayak MK, Banerjee A, Sarkar R, Mitra S, Dutta K, Ganguly N, Ghosh C, Girish Kumar CP, Niyogi P, Panda S, et al. Genetic characterization of group-A rotaviruses among children in eastern India during 2014–2016: phylodynamics of co-circulating genotypes. Vaccine. 2019;37(45):6842–56. doi:10.1016/j.vaccine.2019.06.062.
- World Health Organization. Draft landscape of COVID-19 candidate vaccines. Geneva (Switzerland): World Health Organization; 2020.
- Thanh Le T, Andreadakis Z, Kumar A, Gomez Roman R, Tollefsen S, Saville M, Mayhew S. The COVID-19 vaccine development landscape. Nat Rev Drug Discov. 2020;19(5):305–06. doi:10.1038/d41573-020-00073-5.
- Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, Guo L, Guo R, Chen T, Hu J, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020;11(1):1620. doi:10.1038/s41467-020-15562-9.
- Yin C. Genotyping coronavirus SARS-CoV-2: methods and implications. Genomics. 2020;112:3588–96. doi:10.1016/j.ygeno.2020.04.016.
- Rogers TF, Zhao F, Huang D, Beutler N, Burns A, He WT, Limbo O, Smith C, Song G, Woehl J, et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science. 2020;369:956–63. doi:10.1126/science.abc7520.
- Yuan Y, Wang N, Ou X. Caution should be exercised for the detection of SARS-CoV-2, especially in the elderly. J Med Virol. 2020;92(9):1641–48. doi:10.1002/jmv.25796.
- Xiao AT, Tong YX, Zhang S. False-negative of RT-PCR and prolonged nucleic acid conversion in COVID-19: rather than recurrence. J Med Virol. 2020;92(10):1755–56. doi:10.1002/jmv.25855.
- Xiang F, Wang X, He X, Peng Z, Yang B, Zhang J, Zhou Q, Ye H, Ma Y, Li H, et al. Antibody detection and dynamic characteristics in patients with COVID-19. Clin Infect Dis. 2020;71(8):1930–34. doi:10.1093/cid/ciaa461.
- Peng J, Wang M, Zhang G, Lu E. Seven discharged patients turning positive again for SARS-CoV-2 on quantitative RT-PCR. Am J Infect Control. 2020;48(6):725–26. doi:10.1016/j.ajic.2020.03.017.
- Okba NMA, Muller MA, Li W, Wang C, GeurtsvanKessel CH, Corman VM, Lamers MM, Sikkema RS, de Bruin E, Chandler FD, et al. Severe acute respiratory syndrome coronavirus 2-specific antibody responses in coronavirus disease 2019 patients. Emerg Infect Dis. 2020;26(7):1478–88.
- Diaz-Quijano FA, Rodriguez-Morales AJ, Waldman EA. Translating transmissibility measures into recommendations for coronavirus prevention. Rev Saude Publica. 2020;54:43. doi:10.11606/s1518-8787.2020054002471.
- Ke R, Li H, Wang S, Ding W, Ribeiro RM, EE G, Bhattacharya T, Barnard RJO, Hahn BH, Shaw GM, et al. Superinfection and cure of infected cells as mechanisms for hepatitis C virus adaptation and persistence. Proc Natl Acad Sci U S A. 2018;115:E7139. doi:10.1073/pnas.1805267115.
- Capobianchi MR, Rueca M, Messina F, Giombini E, Carletti F, Colavita F, et al. Molecular characterization of SARS-CoV-2 from the first case of COVID-19 in Italy. Clin Microbiol Infect. 2020;26(7):954–56.
- Beerenwinkel N, Daumer M, Sing T, Rahnenfuhrer J, Lengauer T, Selbig J, Hoffmann D, Kaiser R. Estimating HIV evolutionary pathways and the genetic barrier to drug resistance. J Infect Dis. 2005;191:1953–60. doi:10.1086/430005.
- Beerenwinkel N, Lengauer T, Daumer M, Kaiser R, Walter H, Korn K, Hoffmann D, Selbig J. Methods for optimizing antiviral combination therapies. Bioinformatics. 2003;19(Suppl 1):i16–25. doi:10.1093/bioinformatics/btg1001.
- Andreakos E, Tsiodras S. COVID-19: lambda interferon against viral load and hyperinflammation. EMBO Mol Med. 2020. doi:10.15252/emmm.202012465.
- Diurno F, Numis FG, Porta G, Cirillo F, Maddaluno S, Ragozzino A, De Negri P, Di Gennaro C, Pagano A, Allegorico E, et al. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur Rev Med Pharmacol Sci. 2020;24:4040–47. doi:10.26355/eurrev_202004_20875.
- Abena PM, Decloedt EH, Bottieau E, Suleman F, Adejumo P, Sam-Agudu NA, Muyembe TamFum -J-J, Seydi M, Eholie SP, Mills EJ, et al. Chloroquine and hydroxychloroquine for the prevention or treatment of novel coronavirus disease (COVID-19) in Africa: caution for inappropriate off-label use in healthcare settings. Am J Trop Med Hyg. 2020;102(6):1184–88. doi:10.4269/ajtmh.20-0290.
- Borba MGS, Val FFA, Sampaio VS, Alexandre MAA, Melo GC, Brito M, Mourão MPG, Brito-Sousa JD, Baía-da-Silva D, Guerra MVF, et al. Effect of high vs low doses of chloroquine diphosphate as adjunctive therapy for patients hospitalized with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection: a randomized clinical trial. JAMA Netw Open. 2020;3:e208857. doi:10.1001/jamanetworkopen.2020.8857.
- Costanzo M, De Giglio MAR, Roviello GN. SARS-CoV-2: recent reports on antiviral therapies based on lopinavir/ ritonavir,darunavir/ umifenovir,hydroxychloroquine, remdesivir, favipiravir and other drugs for the treatment of the new coronavirus. Curr Med Chem. 2020. doi:10.2174/0929867327666200416131117.
- Damle B, Vourvahis M, Wang E, Leaney J, Corrigan B. Clinical pharmacology perspectives on the antiviral activity of azithromycin and use in COVID-19. Clin Pharmacol Ther. 2020;108(2):201–11. doi:10.1002/cpt.1857.
- Zhai P, Ding Y, Wu X, Long J, Zhong Y, Li Y. The epidemiology, diagnosis and treatment of COVID-19. Int J Antimicrob Agents. 2020;55(5):105955. doi:10.1016/j.ijantimicag.2020.105955.
- Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30(3):269–71. doi:10.1038/s41422-020-0282-0.
- Felsenstein S, Herbert JA, McNamara PS, Hedrich CM. COVID-19: immunology and treatment options. J Clin Immunol. 2020;215:108448. doi:10.1016/j.clim.2020.108448.
- Frost SDW, Volz EM. Viral phylodynamics and the search for an ‘effective number of infections’. Philos Trans R Soc Lond B Biol Sci. 2010;365:1879–90. doi:10.1098/rstb.2010.0060.
- Meriki HD, Tufon KA, Anong DN, Atanga PN, Anyangwe IA, Cho-Ngwa F, et al. Genetic diversity and antiretroviral resistance-associated mutation profile of treated and naive HIV-1 infected patients from the Northwest and Southwest regions of Cameroon. PLoS One. 2019;14:e0225575. doi:10.1371/journal.pone.0225575.
- Perez-Losada M, Castel AD, Lewis B, Kharfen M, Cartwright CP, Huang B, et al. Characterization of HIV diversity, phylodynamics and drug resistance in Washington, DC. PLoS One. 2017;12:e0185644. doi:10.1371/journal.pone.0185644.
- Rodriguez-Morales AJ, Rodriguez-Morales AG, Méndez CA, Hernández-Botero S. Tracing new clinical manifestations in patients with COVID-19 in Chile and its potential relationship with the SARS-CoV-2 divergence. Curr Trop Med Rep. 2020;18:1–4.
- Sah R, Rodriguez-Morales AJ, Jha R, Chu DKW, Gu H, Peiris M, et al. Complete genome sequence of a 2019 novel coronavirus (SARS-CoV-2) strain isolated in Nepal. Microbiol Resour Announc. 2020:9. doi:10.1128/MRA.00169-20.