
ABSTRACT
Innovative therapies to complement current treatments are needed to curb the growing incidence of fatal overdoses related to synthetic opioids. Murine and chimeric monoclonal antibodies (mAb) specific for fentanyl and its analogs have demonstrated pre-clinical efficacy in preventing and reversing drug-induced toxicity in rodent models. However, mAb-based therapeutics require extensive engineering as well as in vitro and in vivo characterization to advance to first-in-human clinical trials. Here, novel murine anti-fentanyl mAbs were selected for development based on affinity for fentanyl, and efficacy in counteracting the pharmacological effects of fentanyl in mice. Humanization and evaluation of mutations designed to eliminate predicted post-translational modifications resulted in two humanized mAbs that were effective at preventing fentanyl-induced pharmacological effects in rats. These humanized mAbs showed favorable biophysical properties with respect to aggregation and hydrophobicity by chromatography-based assays, and thermostability by dynamic scanning fluorimetry. These results collectively support that the humanized anti-fentanyl mAbs developed herein warrant further clinical development for treatment of fentanyl toxicity.
Introduction
Fatal overdoses involving opioids are at an all-time high in the United States,Citation1 and overdose events involving potent synthetic opioids such as fentanyl and its analogs have increased annually since 2013.Citation2–6 Between February 2021 and February 2022, 69,237 overdose deaths associated with synthetic opioids (excluding methadone) were reported, accounting for 88.5% of all opioid-related overdose deaths and 66.6% of all drug-related overdose deaths in the United States.Citation1,Citation7 The current treatment for reversal of opioid-related overdose toxicity is naloxone, a µ-opioid receptor (MOR) antagonist.Citation8 Naloxone is typically effective for combating opioid overdose; however, for more potent MOR agonists that exhibit a serum half-life longer than that of naloxone (30–90 minutes),Citation9 higher or additional doses of naloxone are often required to reverse overdose and protect against renarcotization. Fentanyl has a serum half-life of 8 hrs;Citation10 consequently, additional dosing of naloxone and extended monitoring for signs of recurring fentanyl toxicity can be needed for 2 or more hours.Citation11,Citation12 Additionally, toxicity related to fentanyl and its analogs is partially mediated by non-MOR signaling such as adrenergic and cholinergic pathways, and therefore refractory to naloxone treatment.Citation13 The drastic and sustained increase in overdose deaths related to synthetic opioids indicates that current methods of intervention are inadequate; therefore, the development of alternative or complementary treatment options is paramount.
Monoclonal antibodies (mAb) represent a promising alternative treatment modality for counteracting toxic doses of small-molecule drugs by sequestration of the target molecules in serum, preventing their distribution to drug receptors in the brain. With a typical serum half-life of 21 days in humans,Citation14,Citation15 mAbs offer a 500-fold increase in duration of protection compared to naloxone. This long serum half-life may provide anti-opioid (α-opioid) mAb-mediated protection against renarcotization from fentanyl and/or other MOR agonists that persists after naloxone is metabolized, with the potential for extended protection against subsequent fentanyl exposure after mAb administration. Opioid overdose reversal treatment with a MOR antagonist like naloxone can have undesirable side effects, including precipitation of opioid withdrawal symptoms.Citation9,Citation11 Meanwhile, because the mechanism of action of an α-opioid mAb is drug sequestration rather than receptor antagonism, precipitated withdrawal is not expected to occur with mAb treatment. Though this has not yet been explored with an α-opioid mAb, a nicotine-specific mAb was shown to not precipitate withdrawal in nicotine dependent rats.Citation16 In addition to protection from and reversal of overdose, α-opioid mAb may provide utility in the prophylactic treatment of patients with opioid use disorder (OUD), as α-opioid vaccines have shown pre-clinical efficacy against various models of OUD.Citation17,Citation18
Several mAbs targeting small molecule drugs of abuse are in various stages of clinical and pre-clinical study. Murine and chimeric anti-fentanyl (α-fentanyl) mAb have shown efficacy in rodent models.Citation19–21 A partially humanized anti-cocaine mAb is in late-stage pre-clinical development.Citation22 A chimeric anti-methamphetamine mAb (IXT-m200) that offers protection against methamphetamine toxicity has shown promising safety data in Phase I clinical trials,Citation23–25 and has completed one Phase II study with a second Phase II study (NCT05034874) in progress. Despite this success with murine and chimeric mAbs (chAb) in pre-clinical and clinical settings, it has been well described that murine mAbs are typically not suitable as a therapeutic treatment in humans due to immunogenicity leading to production of anti-drug antibodies (ADA),Citation26 and chAb with human constant regions and murine variable regions are also susceptible to immunogenicity.Citation27 Human mAb derived from humanized transgenic mouse or rat models are industry-standard in mAb discovery; however, access to such models is often limited by the costly licensing fees associated with their use or by their proprietary ownership. Thus, in vitro humanization of murine-derived mAb remains a validated mAb engineering method for the development of therapeutic mAb candidates with a decreased immunogenicity risk compared to murine mAbs and chAbs;Citation27,Citation28 and to date more than 50 humanized mAbs have been approved by the US Food and Drug Administration (FDA) or the European Medicines Agency.Citation29
This study describes the isolation, humanization, and in vitro and in vivo characterization of novel α-fentanyl mAbs. Murine and chimeric α-fentanyl mAbs prevented fentanyl-induced pharmacological effects and reduced fentanyl distribution to the brain in mice. Two mAbs were selected for humanization via a complementarity-determining region (CDR) grafting approach, evaluated to ensure that binding to fentanyl was maintained, and tested for biophysical properties consistent with mAb candidates suitable for clinical development and manufacturing. These humanized mAbs showed high affinity for fentanyl, displayed favorable biophysical properties, and prevented pharmacological effects induced by 0.3 mg/kg fentanyl in rats.
Materials and methods
Animals
All experiments were approved by the University of Minnesota Animal Care and Use Committee prior to initiation and were conducted according to the Guide for the Care and Use of Laboratory Animals, 8th ed. Male and female Balb/c mice and male Sprague Dawley rats were 8–10 weeks on arrival, were housed in standard conditions with a 14/10 hr light/dark cycle, and provided with food and water ad libitum. Animals were acclimated to the housing environment for 1 week prior to initiation of experiments.
Hybridomas
Isolation of HY6-F9_Mu was previously described.Citation19 To isolate HY11 mAbs, mice (n = 2 male and 2 female) were immunized s.c. with 75 µg F1-CRMCitation18 adsorbed on alum adjuvant (Alhydrogel-85, Invivogen, Catalog # vac-alu-250). Serum was collected via facial vein sampling on day 14 post-immunization. Due to work interruptions related to the COVID-19 pandemic, no additional boosts were performed, and splenocytes were collected and frozen in FBS +7% DMSO on day 18 after initial vaccination. Generation and characterization of fentanyl-specific hybridomas was performed as described.Citation19 Upon confirmation of secretion of fentanyl-specific IgG, sequencing of IgG antibody variable regions was performed as described.Citation30
Generation of chimeric and humanized mAb expression vectors
Fentanyl-binding mAb VH and VL sequences were cloned into pcDNA3.4 mammalian expression vectors prepared by Genscript. The CMV promoter-driven pcDNA3.4 expression vector was modified to contain a Kozak consensus sequence preceding an open-reading frame (ORF) with a murine IGHV signal peptide (MGWSCIILFLVATATGVHS), or a murine IGKV signal peptide (METDTLLLWVLLLWVPGSTG) for the antibody heavy (HC) and light chain (LC) expression vector, respectively. The HC vector ORF terminates with a human IgG1 constant region (Accession # P01857), and the LC vector ORF terminates with a human IgK constant region (Accession # P01834). Both pcDNA3.4 expression vectors were designed with cloning sites between the signal peptide and the constant region to facilitate an in-frame Gibson assembly cloning strategy for variable region insertion with Gibson Assembly® Master Mix (New England Biolabs Catalog # E2611). Inserts for Gibson assembly of chAb expressing plasmids were prepared by variable region PCR amplification of the PCR product generated during the murine mAb VH/VL sequencing procedure. Inserts for Gibson assembly of humanized antibody expressing plasmids were codon optimized and synthesized by Twist Bioscience.
Expression and purification of mAb
Murine mAb was produced by hybridomas cultured in 25–100 mL ClonaCellTM-HY Medium E (Stemcell Technologies Catalog # 03805) depleted of bovine IgG via liquid chromatography on an ÄKTA pure (Cytiva) with a HiTrap Protein G HP column (Cytiva Product # 29048581). Cell culture supernatant was harvested when cell viability fell below 20%, and mAb was purified via liquid chromatography on an ÄKTA pure with a HiTrap Protein G HP column (running buffer PBS, pH 7.4, elution buffer 0.1 M glycine, pH 2.5).
Chimeric and humanized mAb were produced via transient expression with the Expi293 or ExpiCHO expression system according to manufacturer instructions (ThermoFisher Catalog # A14635 and A29133). Cell culture supernatant was harvested 5–10 days following transfection and mAb was purified via liquid chromatography on an ÄKTA pure with a HiTrap MabSelect PrismA protein A column (Cytiva Product # 17549851) (running buffer PBS, pH 7.4, elution buffer 0.1 M Na-Acetate, pH 3.5).
For both hybridoma and transiently produced mAb, eluted mAb was neutralized by dilution with 1/3rd final volume 2.5 M Tris, pH 7.2, and buffer exchanged into PBS, pH 7.4. Purified mAb concentration was determined by absorbance at 280 nm on a Nanodrop. Confirmatory analysis of purified mAb was performed by SDS-PAGE under reducing and non-reducing conditions.
In-house control rituximab was generated using the human IgG1 and IgK pcDNA3.4 expression vectors and the ExpiCHO expression and Protein A purification procedure described above. The VH and VL sequence of rituximab was obtained from go.drugbank.com, Accession Number DB00073.
Determination of antibody titer, affinity, and selectivity
Relative affinity of mAb to various compounds was performed by competitive ELISA with F3-BSA as the coating antigen as previously described.Citation18,Citation19 Estimated affinity was quantitated as IC50, or concentration of competitor that reduced antibody binding to plates by 50%, and % relative affinity was expressed as (fentanyl IC50)/(competitor IC50)*100. Antibody titer in hybridoma supernatant was determined using an Octet Red96e (Sartorius). Protein G biosensors (Sartorius Catalog # 18–5082) were pre-incubated in conditioned medium from Sp2/0 cells grown in ClonaCellTM-HY Medium E depleted of bovine IgG for 60 sec. Next, biosensors were incubated in hybridoma supernatant for 60 sec and the association between the Protein G biosensor and murine IgG in supernatant was measured and quantitated against a standard curve of purified murine IgG1 resuspended in conditioned medium. All calculations were performed with Octet analysis software (Sartorius). Antibody titer determination of transfected Expi293 or ExpiCHO supernatant was performed as described above, but with Protein G biosensor pre-incubation in conditioned medium from untransfected Expi293 or ExpiCHO cells and the standard curve was generated with purified human IgG1.
Affinity and antigen selectivity measurements by BLI were performed with an Octet Red96e with biotinylated haptens derived from fentanyl (F1),Citation18,Citation31 acetylfentanyl (F10),Citation31 and carfentanil (F11).Citation32 Streptavidin-coated biosensors (Sartorius Catalog # 18–5020) were pre-incubated in 10 mM sodium phosphate, 0.15 M NaCl, 0.05% Tween-20, pH 7.5 (PBS-T) and loaded with 0.2 µg/mL of biotinylated hapten for 60 sec. After a 60 sec baseline measurement in PBS-T, association of 1–100 nM mAb with hapten-biotin was measured for 3–5 min, followed by dissociation measurement in PBS-T for 5–10 min. The Octet analysis software (Sartorius) performed all calculations of on-rate (kon), off-rate (koff), and KD (koff/kon). Full kon, koff, and KD for each mAb is available in Table S3.
Selection of human VH and VL germline gene for humanization by CDR grafting
For both the VH and VL chain, three human germline gene sequences with the highest homology and/or Smith-Waterman score, as reported by the DomainGapAlign webtool,Citation33 were chosen for in silico characterization and developability assessment with TAP: Therapeutic Antibody Profiler to determine if any chosen germline gene sequences harbored developability risks.Citation34 Based on homology/Smith-Waterman score from DomainGapAlign and in silico characterization results from TAP, a final human VH or VL germline gene sequence was chosen for humanization by CDR grafting.
Post-translational modification (PTM) mitigation
CDR amino acid residues susceptible to deamidation, glycosylation, and isomerization were identified with the abYsis webtool.Citation35 Methionine and tryptophan oxidation were not considered for PTM mitigation. In VL CDR1 of HY6-F9_Hu, Asn34 (IMGT numbering) is followed by Gly, forming the highly susceptible asparagine deamidation motif, “NG” (Table S1).Citation36,Citation37 Three preemptive PTM mitigation mutations were introduced into VL CDR1 of HY6-F9_Hu: N34Q, G35K, and G35 R. Additionally, to determine if deamidation of N34 would affect binding to the fentanyl hapten, a N34D mutation was introduced to mimic deamidation of N34.Citation37
In the VH CDR2 of HY11-7E1_Hu, an Asp62 (IMGT numbering) is followed by Gly, forming the highly susceptible aspartate isomerization motif, “DG” (Table S1).Citation38 Two preemptive PTM mitigation mutations were introduced into VH CDR2 of HY11-7E1_Hu: D62E, and G63 V. The G63 V mutation was chosen based on the “N + 1” strategy for aspartate isomerization mitigation.Citation39
Point mutations were introduced via site-directed mutagenesis with a QuikChange Multi Site-Directed Mutagenesis Kit (Agilent Catalog # 200514) and mutations were confirmed by Sanger sequencing.
Biophysical characterization – mAb aggregation and fragmentation analysis by SEC-HPLC
The relative amount of aggregate, monomer, and fragment in purified mAb sample was determined by SEC-HPLC on an Agilent 1200 series HPLC with a quaternary pump and multi-wave detector. Purified mAb (20 µg) was run on a Shodex KW-803 column preceded by a Shodex KW-G guard column.
SEC-HPLC method details: mobile phase = PBS (isocratic), pH 7.0, flow rate = 0.75 mL/min, sample injection = 20 µg, run time = 30 min, operating temperature = 24°C, detection = absorbance at 280 nm. Integration of the chromatogram was performed with ChemStation software (Agilent), using system standard settings for “new exponential.” Peaks eluting off the column before the main peak were assessed as aggregate. Peaks eluting off the column after the main peak were assessed as fragment. All SEC-HPLC runs were performed by the Biotechnology Resource Center of the BioTechnology Institute at the University of Minnesota.
Biophysical characterization – mAb hydrophobicity analysis by HIC-HPLC
The relative hydrophobicity of purified mAb was determined by HIC-HPLC on an Agilent 1200 series HPLC with a quaternary pump and multi-wave detector. Purified mAb was run on a MAbPac HIC-10, 4.6x100 column (ThermoFisher Catalog # 088480) preceded by a MAbPac HIC-10, 4.6x10 guard column (ThermoFisher Catalog # 088482).
HIC-HPLC method details: mobile phase A = 100 mM sodium phosphate, 1.5 M ammonium sulfate, pH 7.0, mobile phase B = 100 mM sodium phosphate, pH 7.0, flow rate = 0.75 mL/min, operating temperature = 25°C, detection = absorbance at 280 nm. The HPLC method proceeded as follows: (1) Equilibration; 100% mobile phase A, −3.0 to 0.0 min. (2) Sample injection; 25 µg of purified mAb diluted 1:1 with mobile phase A. (3) Gradient phase; 100% mobile phase A to 100% mobile phase B, 0 to 15.0 min. (4) Column flush; 100% mobile phase B, 15.0 to 18.0 min. (5) Shutdown, 100% mobile phase A, 18.0 to 19.0 min. Integration of the chromatogram was performed with ChemStation, using system standard settings for “new exponential.” The manufacturer specified void volume of the MAbPac HIC-10, 4.6x100 column is 1.25 mL, or 1.67 minutes when operating at a flow rate of 0.75 mL/min. All HIC-HPLC runs were performed by the Biotechnology Resource Center of the BioTechnology Institute at the University of Minnesota.
Biophysical characterization – Fab Tm determination by DSF
The Tm of the Fab fragment of purified mAb was determined by DSF with a StepOnePlus™ Real-Time PCR System (Applied Biosystems Catalog # 4376600). For the measurement, mAb at 1 mg/mL in PBS, pH 7.4 was combined with the assay reagents from the Protein Thermal ShiftTM Dye Kit (Applied Biosystems Catalog # 4461146) to a final volume of 20 µL and subjected to a continuous 0.3% temperature ramp from 25 to 95°C. Fluorescence measurements were recorded during the temperature ramp. Fab Tm determination was performed with Protein Thermal Shift™ Software v1.4 (Applied Biosystems Catalog # 4466038). Reported Tm values represent the mean of four sample replicates.
Efficacy of mAb against fentanyl in vivo
Animals were acclimated to the testing environment for 1 hr prior to drug challenge. For determination of mAb efficacy against fentanyl, mice were passively immunized s.c. with 40 mg/kg mAb (molar equivalent of 0.15–0.2 mg/kg fentanyl). 24 hours post-immunization, mice were challenged with 0.1–0.25 mg/kg fentanyl s.c. as indicated in figure legends. Rats were passively immunized with 40 mg/kg mAb i.p. and challenged with 0.1–0.3 mg/kg fentanyl s.c. At 15-minute intervals post-challenge, animals were evaluated for drug-induced antinociception by hot plate set to 54°C (Columbus Instruments), and for drug-induced respiratory depression and bradycardia with MouseOx Plus pulse oximeter (Starr Life Science). Following the final behavioral measurement, animals were euthanized by CO2 inhalation, and brain and serum were collected for analysis of fentanyl and norfentanyl concentration by LCMS as described.Citation18,Citation19 Animals were randomly assigned to control or mAb groups by body weight, and antinociception and oximetry measurements were taken by experimenters blinded to treatment condition. Differences between groups in bradycardia, respiratory depression and antinociception were evaluated by 2-way ANOVA followed by Dunnett’s or Tukey’s multiple comparisons post-hoc test. Analyses were conducted in Prism v9.2 (GraphPad).
Results
Characterization of murine α-fentanyl mAb
Anti-fentanyl murine mAbs were isolated from hybridomas generated from splenocytes of mice immunized with fentanyl vaccine F1-CRM.Citation18 Hybridoma clones were screened for binding to fentanyl hapten by ELISA (data not shown), and positive clones were advanced for in vitro and in vivo characterization. Six murine mAbs that bound to fentanyl were identified: HY6-F9_Mu,Citation19 HY11-2F5_Mu, HY11-4E6_Mu, HY11-5C1_Mu, HY11-6B2_Mu, and HY11-7E1_Mu. To determine KD and assess cross-reactivity of novel α-fentanyl murine mAbs to fentanyl analogs, binding of purified mAb to biotinylated haptens derived from fentanyl, acetylfentanyl, and carfentanil was measured by biolayer interferometry (BLI). All mAbs had measured KD values of <0.1 nM to the fentanyl hapten (), while all HY11 mAbs also showed KD values of <0.1 nM to the acetylfentanyl hapten, with HY6-F9_Mu yielding a KD of 0.63 nM to the acetylfentanyl hapten. Only HY11-2F5_Mu and HY11-7E1_Mu showed measurable binding (KD = 0.72 nM and 1.18 nM, respectively) for the carfentanil hapten.
Table 1. KD of murine, chimeric and humanized mAb determined by biolayer interferometry (BLI).
The relative affinity of lead mAbs for fentanyl compared to non-target compounds was evaluated by competitive ELISA. All mAbs showed nanomolar relative affinity (IC50) for fentanyl and acetylfentanyl; and for norfentanyl, a fentanyl metabolite (), and only HY11-7E1_Mu showed binding to carfentanil (KD = <1 µM). By contrast, mAbs showed little to no binding to off-target opioid receptor ligands including buprenorphine, methadone, morphine, oxycodone, naloxone, naltrexone and tramadol, or to other non-opioid off-target drugs including methamphetamine, nicotine and common over-the-counter drugs acetaminophen, aspirin, or ibuprofen. The high specificity of mAb for fentanyl and its derivatives supports that these mAb should not interfere with commonly encountered compounds if present in a clinical setting.
Table 2. Relative affinity of mAb for fentanyl and other target and off-target compounds as determined by competitive ELISA.
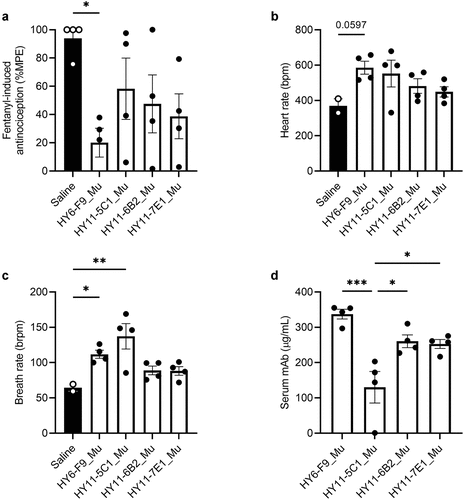
To determine whether isolated mAbs were effective at reducing fentanyl effects in vivo, mice were passively immunized with 40 mg/kg of HY6-F9_Mu, HY11-5C1_Mu, HY11-6B2_Mu, and HY11-7E1_Mu, and challenged with 0.25 mg/kg fentanyl s.c. (). HY11-2F5_Mu was not chosen for in vivo testing due to sub-optimal levels of aggregation as measured by size-exclusion high pressure liquid chromatography (SEC-HPLC) (data not shown), and HY11-4E6_Mu was not chosen due to high sequence similarity to HY6-F9_Mu. Treatment with mAb somewhat reduced fentanyl-induced antinociception, though only the effect of HY6-F9 was significant (); and HY6-F9_Mu and HY11-5C1_Mu increased breath rate 30 minutes after fentanyl challenge relative to saline control (). One week after fentanyl challenge, concentration of mAb in serum was measured (); the serum level of mAb in mice treated with HY11-5C1_Mu was significantly decreased compared to other mAbs, indicating this mAb may have lower serum stability than the other lead mAbs.
Figure 1. In vivo efficacy against fentanyl of murine mAbs. Mice (n=4 per group, 2 males and 2 females) were passively immunized with anti-fentanyl mAb (40 mg/kg, s.c.), and 24 hours later were challenged with 0.25 mg/kg fentanyl. Fentanyl-induced effects on: (a) antinociception measured by hot plate; (b) heart rate and (c) breath rate measured by pulse oximetry. (d) One week after challenge, serum concentration of mAb measured by ELISA. Data are expressed as mean ± SEM; *p≤.05; **p≤.01; ***p≤.001.
Characterization and efficacy of chimeric α-fentanyl mAb
Heavy chain variable region (VH) and light chain variable region (VL) sequences from the lead murine mAbs HY6-F9_Mu, HY11-6B2_Mu, and HY11-7E1_Mu were cloned into expression vectors with human IgG1 and IgK constant region sequences to generate chAbs. Purified chAbs were assessed for binding to biotinylated fentanyl, acetylfentanyl, and carfentanil haptens by BLI (). All chAbs maintained KD values below 0.1 nM for fentanyl hapten, matching their murine counterpart. The HY6-F9 and HY11-6B2 chAbs (HY6-F9_Ch and HY11-6B2_Ch) bound to the acetylfentanyl hapten with comparable binding affinities to their murine counterpart, while the affinity of chimeric HY11-7E1 (HY11-7E1_Ch) to acetylfentanyl hapten decreased (<0.1 nM to 0.28 nM). HY11-7E1_Ch maintained binding to the carfentanil hapten (0.51 nM).
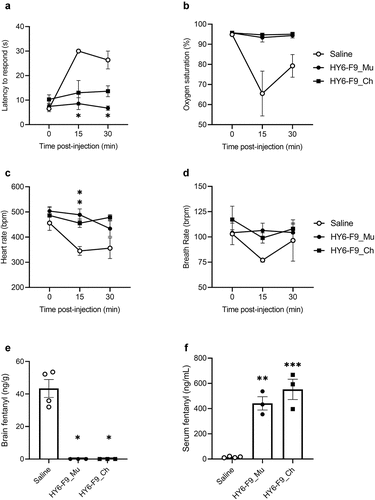
To evaluate whether in vivo efficacy of chAb was maintained, mice were passively immunized with 40 mg/kg of each murine mAb or chAb and challenged with 0.1 mg/kg fentanyl (). Concentrations of fentanyl in brain and serum were measured 30 min after fentanyl administration. All mAbs significantly reduced brain fentanyl and increased fentanyl concentration in serum, though HY11-6B2_Ch was least effective at sequestering fentanyl in serum. Because HY6-F9_Mu has previously shown efficacy in rats,Citation19,Citation40 the efficacy of HY6-F9_Ch was also compared to HY6-F9_Mu in rats (). Rats were passively immunized with 40 mg/kg of HY6-F9_Mu or HY6-F9_Ch, and 24 hours later challenged with 0.1 mg/kg fentanyl. Both HY6-F9_Mu and HY6-F9_Ch prevented fentanyl-induced antinociception (), respiratory depression and bradycardia (), and reduced fentanyl distribution to brain (). These results and the ability of HY11-7E1 to bind carfentanil supported the selection of HY6-F9 and HY11-7E1 as lead mAbs for humanization and further development.
Figure 2. In vivo comparison of murine and chimeric anti-fentanyl mAbs. Mice (n=3 male mice per group) were passively immunized with anti-fentanyl mAb (40 mg/kg, s.c.), and 24 hours later were challenged with 0.1 mg/kg fentanyl. Concentration of fentanyl in (a) brain and (b) serum measured by LCMS. (c) Serum concentration of mAb and chAb prior to fentanyl challenge measured by ELISA. Data are expressed as mean ± SEM; *p≤.05; ***p≤.001, ****p≤.0001.
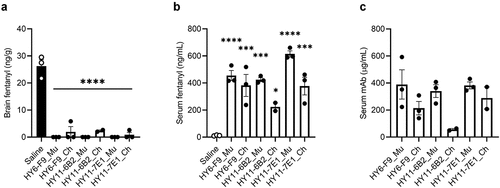
Figure 3. In vivo comparison of murine and chimeric HY6-F9 in rats. Rats (n=3–4 male rats per group) were passively immunized with anti-fentanyl mAb (40 mg/kg, i.p.), and 24 hours later were challenged with 0.1 mg/kg fentanyl. (a) Fentanyl-induced antinociception as latency to respond on a hot plate; (b) oxygen saturation, (c) heart rate, and (d) breath rate measured by pulse oximetry. Concentrations of fentanyl in (e) brain and (f) serum measured by LCMS. Data are expressed as mean ± SEM; *p≤.05; **p≤.01; ***p≤.001.
Humanization of α-fentanyl mAb
To reduce potential immunogenicity in response to murine VH and VL regions present on the chAbs,Citation27,Citation41,Citation42 CDR amino acids of HY6-F9_Ch and HY11-7E1_Ch were grafted onto human germline VH and VL gene sequences chosen as described in the Materials and Methods. CDR grafting was accomplished using two grafting strategies: CDR amino acid regions delineated by (1) IMGT definitions,Citation43 or (2) combined IMGT, KABAT,Citation44 CHOTHIA,Citation45 AbM,Citation46 and paratome definitions.Citation47 Because grafting strategy 1 encompasses fewer murine CDR amino acids relative to grafting strategy 2, humanized mAb grafted with only the IMGT defined CDR amino acids resulted in the highest % homology for human sequence; therefore, this grafting scheme was denoted “high homology.” For grafting strategy 2, considering that the combined IMGT, KABAT, CHOTHIA, AbM, and paratome definitions encompassed a larger number of murine CDR amino acids relative to grafting strategy 1, this grafting scheme was denoted “low homology.” To isolate any loss in binding to either the humanized VH or VL, humanization was conducted with a stepwise approach. First, high and low homology humanized VH were paired with chimeric VL to produce humanized intermediates. These humanized intermediates were evaluated for binding to fentanyl and carfentanil by BLI (). Then, high and low homology VL were paired with the humanized VH that retained highest affinity for fentanyl hapten to produce fully humanized mAbs, which were evaluated for fentanyl and carfentanil binding by BLI ().
Table 3. Homology of mAbs to human germline and KD to fentanyl and carfentanil.
The amino acid sequence % homology of the hybridoma-derived murine IgG1/IgK α-fentanyl mAb to a human IgG1/IgK mAb containing the corresponding germline gene VH/VL sequences used for humanization was 67.8% for HY6-F9_Mu and 64.9% for HY11-7E1_Mu (). For HY6-F9, the humanized mAb with low homology VH and VL displayed a 50-fold increase in KD for fentanyl hapten by BLI compared to humanized mAb containing low homology VH and high homology VL (). For HY11-7E1, both low and high homology humanized VH intermediate mAb showed similar binding to fentanyl and carfentanil haptens. However, when fully humanized, the humanized mAb containing the high homology VL showed a marked reduction in affinity for fentanyl hapten, and binding to carfentanil hapten was ablated (). The humanized α-fentanyl mAbs chosen for further development, HY6-F9_Hu and HY11-7E1_Hu, contained low homology VH and VL, resulting in 96.7% and 95.0% homology to human IgG1/IgK mAb containing the corresponding germline gene VH/VL, respectively.
PTM mitigation of humanized α-fentanyl mAb
Amino acids residues in the combined KABAT + IMGT defined CDR regions of HY11-7E1_Hu and HY6-F9_Hu prone to PTM were identified and modified to reduce the risk of PTM-induced mAb heterogeneity and immunogenicity.Citation48 One potential PTM risk was identified in the CDRs of each mAb, and specific details on the mitigation and mimicking strategy can be found in the Materials and Methods. To determine whether the introduction of PTM mitigating or mimicking mutations at these residues would impact affinity for fentanyl, mAbs incorporating these mutations were produced and evaluated for binding to hapten-biotin by BLI. The PTM mitigated humanized HY6-F9 mAbs (HY6-F9_Hu (NQ), HY6-F9_Hu (GK), HY6-F9_Hu (GR)) and HY11-7E1 mAbs (HY11-7E1_Hu (DE), HY11-7E1_Hu (GV)) all maintained KD values comparable to the unmitigated humanized counterpart (Table S1). The HY6-F9_Hu deamidation mimic (HY6-F9_Hu (ND)) resulted in a 10-fold increase in KD (Table S1), indicating that deamidation of this asparagine in HY6-F9_Hu may decrease efficacy against fentanyl in vivo.
Biophysical characterization of humanized and PTM-mitigated α-fentanyl mAb
Biophysical characterization and developability assessments of candidate α-fentanyl mAbs were performed to identify potential developability liabilities. When considering the selection of a lead mAb candidate for clinical development, biophysical characterization provides further criteria with which to rank mAb candidates against one another based on key characteristics for predicting manufacturing process development success.
Chimeric, humanized, and PTM-mitigated humanized HY6-F9 and HY11-7E1 α-fentanyl mAbs were transiently expressed in CHO cells to approximate the cell line to be used for future stable cell line development and formulated in phosphate buffered saline (PBS), pH 7.4 at 1.0 mg/mL. While PBS, pH 7.4 is not an optimized buffer formulation for a mAb therapeutic, it was chosen for preliminary characterization activities as a baseline for comparison between candidate mAbs. Aggregation by SEC-HPLC,Citation49 hydrophobicity by hydrophobic interaction high pressure liquid chromatography (HIC-HPLC),Citation50 and Fab domain melting temperature (Tm) by dynamic scanning fluorimetry (DSF)Citation51,Citation52 were assessed for all candidate α-fentanyl mAbs (). A commercially approved therapeutic mAb (rituximab) was produced internally to provide a comparator for SEC-HPLC and HIC-HPLC analysis.
Table 4. Biophysical characterization of chimeric and humanized mAb.
Aggregation analysis by SEC-HPLC
Monoclonal antibodies prone to aggregation may display low expression, precipitation, reduced stability, and/or other deleterious properties for a potential therapeutic candidate.Citation53 In a study of 152 human or humanized mAbs, analysis by SEC-HPLC showed that 72% of mAbs displayed >95% monomer, and 89% showed >90% monomer.Citation54 Another study showed 20 out of 21 FDA-approved therapeutic mAbs displayed >97.5% monomer.Citation55
For the mAbs analyzed in this study, all humanized versions of the HY6-F9 and HY11-7E1 mAbs displayed >99.5% monomer by SEC-HPLC (). HY6-F9_Ch and HY11-7E1_Ch displayed 98% and 98.5% monomer respectively, and the control in-house rituximab displayed 99.2% monomer. When previously described in ref.Citation55 rituximab displayed 99.0% monomer by SEC-HPLC.
Hydrophobicity analysis by HIC-HPLC
Monoclonal antibodies with high surface hydrophobicity are prone to aggregation, nonspecific binding and self-interaction, substandard concentratability, and high viscosity.Citation54 To assess hydrophobicity by HIC-HPLC, column retention time of the antibody under a high-to-low salt gradient was measured. Under the conditions used in this experiment, mAbs with high hydrophobicity will display a retention time of ≥16.67 min, as that marks the transition to the salt-free mobile phase in which mAb is no longer subject to a salting-out effect. In reported HIC-HPLC assays performed on 32 FDA-approved therapeutic mAbs and mAbs in early-to-late stage clinical trials, 27 mAbs eluted prior to the transition to a salt-free mobile phase.Citation56 For the mAbs analyzed in this study, all HY11-7E1 mAbs displayed similar retention times, between 13.0 and 13.1 min (). The retention times of the HY6-F9 mAbs were more varied, with HY6-F9_Hu mAb having the longest retention time (therefore highest hydrophobicity) at 16.17 min, and HY6-F9_Hu (NQ) eluting at 14.51 min. The HIC-HPLC results for the candidate α-fentanyl mAbs indicate that the HY6-F9 series of mAbs show longer retention times compared to the HY11-7E1 mAbs. Additionally, the HY11-7E1 series of mAbs all had lower hydrophobicity than the rituximab control ().
Tm analysis by DSF
Monoclonal antibodies that contain Fab domains with a Tm less than 65°C may have conformational stability liabilities,Citation54,Citation57 and are susceptible to instability under stressed conditions.Citation58 These liabilities introduce complexities and greater expense in manufacturing, in addition to complications with respect to long-term storage of mAb drug product.
Figure 4. Fab Tm comparison of chimeric and humanized mAb. HY6-F9 mAbs (a) and HY11-7E1 mAbs (b) at 1 mg/ml in PBS, pH 7.4 were combined with Protein Thermal ShiftTM assay reagents and subjected to a continuous 0.3% (0.45°C/min) temperature ramp from 25 to 95°C. The Tm of each mAb fragment is determined by the temperature measurement at the derivative peak (dPeak). The initial, broad dPeak in each sample corresponds to the CH2domain, while the second dPeak in each sample corresponds to the Fab domain. All humanized HY6-F9 and HY11-7E1 mAbs show an increased temperature shift in Fab Tm upon humanization, indicating increased thermal stability compared to the murine chimeric counterpart.
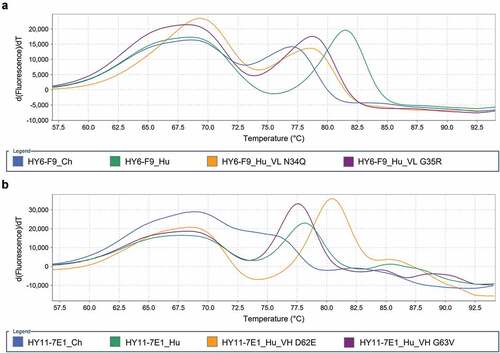
The Fab, CH2, and CH3 domains of IgG typically have distinct unfolding transitions;Citation59 however the Fab domain unfolding transition may overlap with the unfolding transition of the CH2 domain. Thermal stabilization of the Fab domain of an α-cocaine mAb was previously shown to occur upon binding to its small molecule ligand, resulting in a Tm increase of the Fab domain in the presence of cocaine.Citation60 Therefore, Tm was determined in the absence and presence of fentanyl (Supplemental Information, Figure S1, Table S2) to differentiate Fab Tm from the Tm of the CH2 domain. To assess Fab domain Tm of each antibody by DSF, the temperature corresponding to thermal unfolding of the Fab domain, marked by the derivative fluoresence peak, was measured ().
HY6-F9_Hu showed increased Fab thermostability compared to the HY6-F9_Ch, with a ΔTm of 4.5°C (). The PTM mitigated HY6-F9 mAbs had lower thermostability than HY6-F9_Hu, but displayed an overall increased Fab Tm compared to HY6-F9_Ch. The HY11-7E1 mAb with the highest Fab thermostability was HY11-7E1_Hu (DE). The Fab domain of HY11-7E1_Ch showed an unfolding transition that overlapped with the unfolding transition of the CH2; therefore, a discrete Tm value was not captured, and a range of 72.5–76.5°C was estimated. As was the case for the HY6-F9 mAbs, the humanized HY11-7E1 mAbs all displayed improved thermostability compared to their murine and chimeric counterpart. Notably, all mAb showed an increase in Fab Tm in the presence of fentanyl (Supplemental Information, Figure S1, Table S2). In Fab Tm determination assays on 137 FDA approved therapeutic mAbs and mAbs in 2nd or 3rd phase clinical trials, 117 mAbs displayed Fab Tm values ≥65°C when tested under DSF conditions similar to those used in this study, with a mean of 71.3 °C;Citation61 all candidate α-fentanyl mAbs displayed Fab Tm values above the mean Fab Tm value from their study (Figure S2).
Efficacy of humanized lead α-fentanyl mAb
Two humanized α-fentanyl mAbs, HY6-F9_Hu (NQ) and HY11-7E1_Hu (DE), were selected as leads for further evaluation of in vivo efficacy. HY6-F9_Hu (NQ) was chosen as the HY6-F9 lead as the asparagine deamidation mimicking HY6-F9_Hu (ND) mAb showed decreased binding to fentanyl (Table S1), indicating that any post-translational deamidation of N34 in HY6-F9_Hu may decrease mAb efficacy. HY6-F9_Hu (NQ) also displayed a decreased apparent hydrophobicity compared to HY6-F9_Hu (). HY11-7E1_Hu (DE) was chosen as the HY11-7E1 lead due to its apparent higher affinity to fentanyl hapten compared to HY11-7E1_Hu (), its superior Fab Tm value relative to other HY11-7E1 mAbs (), and due to the presence of the aspartate isomerization mitigating VH D62E mutation.
Rats were passively immunized with 40 mg/kg of HY6-F9_Ch, HY6-F9_Hu (NQ), or HY11-7E1_Hu (DE). 24 hours later, rats were challenged with cumulative doses of fentanyl (0.1–0.3 mg/kg), and monitored for fentanyl-induced antinociception, respiratory depression, and bradycardia (). All mAbs significantly reduced fentanyl-induced antinociception up to 0.25 mg/kg fentanyl, and prevented reduction in oxygen saturation and heart rate compared to saline control. At the highest dose of 0.3 mg/kg fentanyl, rats treated with HY11-7E1_Hu (DE) showed a slight reduction in oxygen saturation to approximately 86%, and rats treated with HY6-F9_Hu (NQ) showed significantly higher protection at that dose compared to HY11-7E1_Hu (DE) (p = .043). Finally, while all three mAbs sequestered fentanyl in serum and significantly reduced brain concentration of fentanyl (), HY11-7E1_Hu (DE) was less effective than HY6-F9_Ch or HY6-F9_Hu (NQ) at preventing brain distribution. The concentration of norfentanyl was significantly increased in serum of all passively immunized rats (); brain levels of norfentanyl were below the limit of quantitation (data not shown).
Figure 5. Efficacy of humanized lead mAb in rats. Rats (n=4 male rats per group) were passively immunized with saline, HY6-F9_Ch as positive control, HY6-F9_Hu (NQ), or HY11-7E1_Hu (DE) (40 mg/kg, i.p.), and 24 hours later were challenged with cumulative fentanyl doses up to 0.3 mg/kg. (a) Fentanyl-induced antinociception as latency to respond on a hot plate; (b) oxygen saturation, and (c) heart rate measured by pulse oximetry. (d) Concentration of mAb 1 hour prior to challenge measured by ELISA; and concentrations of fentanyl in (e) serum and (f) brain, and (g) concentration of norfentanyl in serum 15 min after final fentanyl dose measured by LCMS. Data are expressed as mean ± SEM; *, # or † indicate significance of indicated groups vs saline, or bars to indicate significance between groups; [*,#,†]p≤.05; [**,##,††]p≤.01; [***,###,†††]p≤.001; [****,####,††††]p≤.0001.
![Figure 5. Efficacy of humanized lead mAb in rats. Rats (n=4 male rats per group) were passively immunized with saline, HY6-F9_Ch as positive control, HY6-F9_Hu (NQ), or HY11-7E1_Hu (DE) (40 mg/kg, i.p.), and 24 hours later were challenged with cumulative fentanyl doses up to 0.3 mg/kg. (a) Fentanyl-induced antinociception as latency to respond on a hot plate; (b) oxygen saturation, and (c) heart rate measured by pulse oximetry. (d) Concentration of mAb 1 hour prior to challenge measured by ELISA; and concentrations of fentanyl in (e) serum and (f) brain, and (g) concentration of norfentanyl in serum 15 min after final fentanyl dose measured by LCMS. Data are expressed as mean ± SEM; *, # or † indicate significance of indicated groups vs saline, or bars to indicate significance between groups; [*,#,†]p≤.05; [**,##,††]p≤.01; [***,###,†††]p≤.001; [****,####,††††]p≤.0001.](/cms/asset/3ed5e2ec-aac2-4562-991a-acef3b5effeb/khvi_a_2122507_f0005_oc.jpg)
Discussion
The present-day record high overdose death counts involving synthetic opioids are an attestation that current methods of prevention and therapeutic intervention against opioid-related overdose deaths are insufficient. Potentially exacerbating the overdose death rate is that naloxone, the current standard therapeutic intervention to reverse opioid toxicity in overdose scenarios, may be less effective at counteracting fentanyl compared to other opioids.Citation62,Citation63 To address the increased prevalence of synthetic opioids such as fentanyl, mAb treatments that specifically counteract synthetic opioid toxicity are a promising area of study. Anti-fentanyl murine mAb and chAb isolated by other groups have been shown to prevent fentanyl-induced antinociception and brain distribution at similar mAb and fentanyl doses to those used here.Citation20,Citation21 Meanwhile, HY6-F9_Mu has been shown to prevent fentanyl effects in both mice and rats,Citation19 and to reverse respiratory depression in rats,Citation40 a more clinically relevant measure for fentanyl overdose than antinociception. However, as murine and chimeric mAbs are unsuitable for human use due to ADA responses,Citation26–28 humanization is essential for clinical advancement. Hence, engineering and biophysical characterization was conducted for HY6-F9_Mu and the novel mAb HY11-7E1_Mu, yielding humanized and PTM mitigated mAbs that showed <0.25 nM binding to fentanyl, favorable biophysical properties, and no loss of efficacy in rats compared to murine mAb.
The in vitro biophysical characterization assays performed on the lead mAbs described here provide evidence that these mAbs are suitable for further clinical development as therapeutic candidates, as they displayed minimal aggregation and fragmentation, low hydrophobicity, and high Fab thermostability when analyzed by SEC-HPLC, HIC-HPLC, and DSF, respectively. Additionally, any alteration or heterogeneity induced by PTMs in mAb CDRs, such as asparagine deamidation and aspartate isomerization, pose a risk of loss of antibody efficacy.Citation64,Citation65 Where such risks were present in mAb CDRs, PTM mitigating mutations were introduced. PTM mitigated humanized mAbs retained equivalent affinity to fentanyl and carfentanil by BLI (), and they maintained optimal biophysical characteristics (). Combined, these characterization and PTM mitigation results indicate that the candidate α-fentanyl mAbs developed in this study are aligned with currently approved mAb therapeutics.
Upon completion of successful engineering and characterization, two lead humanized mAbs containing PTM mitigating mutations were assessed for in vivo efficacy against fentanyl in rats. These mAbs protected against effects of fentanyl-induced antinociception, respiratory depression, and bradycardia by preventing the distribution of fentanyl to the brain. Importantly, the lead HY6-F9_Hu (NQ) was significantly more effective at sequestering fentanyl in serum compared to HY11-7E1_Hu (DE). Additionally, while HY6-F9_Hu (NQ) prevented respiratory effects of fentanyl (i.e., oxygen saturation (SaO2) >95%) up to 0.3 mg/kg, the protection afforded by HY11-7E1_Hu (DE) was overcome at the higher doses, with SaO2 significantly lower in these rats than in rats treated with HY6-F9_Hu (NQ). While this difference in efficacy cannot be explained by affinity or serum level of the two mAbs, it is possible that HY11-7E1_Hu (DE) does not exhibit optimal fentanyl binding under in vivo conditions compared to in vitro binding assays. The concentration of norfentanyl was also increased in serum of mAb-treated rats, suggesting that metabolism of fentanyl to norfentanyl is not ablated by mAb binding, though the overall effect of mAb on fentanyl metabolism and elimination remains to be fully explored.
Additional characterization activities are necessary to prepare lead α-fentanyl mAbs for the clinic, and to address potential limitations of mAb treatments against synthetic opioid toxicity. For the lead α-fentanyl mAbs developed in this study, in silico modeling as well as in vitro and in vivo assays to predict immunogenicity need to be performed. Forced oxidation, deamidation, and isomerization studies should be performed to assess potential deleterious PTMs in any remaining unmitigated amino acid residues in the CDRs, residues implicated in neonatal Fc receptor (FcRn) binding, and residues that may affect antibody stability.Citation66–68 The current study highlights that further mAb dosing studies in animal models are required to determine dosing, delivery, and formulation strategies for clinical use, as 40 mg/kg of HY6-F9_Hu (NQ) was efficacious against doses up to 0.3 mg/kg fentanyl in rats (equivalent to 12x the lethal dose in humans), while 40 mg/kg of HY11-7E1_Hu (DE) began losing efficacy above 0.25 mg/kg fentanyl in rats. If high mAb doses commonly delivered via i.v. administration are necessary,Citation69–71 development of high concentration mAb formulations,Citation72–74 and/or formulations including hyaluronidase may enable s.c. administration.Citation75
Considering next-generation α-fentanyl mAbs, any limitations resulting from potentially high dosing requirements may be overcome by introducing mAb half-life extension mutations,Citation76 and through recombinant formats that reduce mAb molecular weight per fentanyl binding site. For example, a single-chain variable fragment Fc-fusion (100,000 g/mol vs 150,000 g/mol for a standard mAb) specific to methamphetamine has been developed that displays efficacy against the psychostimulant effects of methamphetamine.Citation77 Furthermore, because instances of exposure to multiple opioids are frequent,Citation78,Citation79 treatment with mAbs that bind to structurally distinct opioids may be desirable. Both lead mAbs showed affinity for acetylfentanyl, HY11-7E1 showed affinity for carfentanil, and a previous mAb has shown in vivo efficacy against carfentanil in mice;Citation21 however, methods for evaluating the ability of a cross-reactive mAb to counteract polydrug exposure need to be developed. Inter-drug competition for the mAb binding pocket will be heavily influenced by each drug’s KD and Kon, and their concentration and relative potency will impact the ability of a cross-reactive mAb to prevent a pharmacologically meaningful amount of each drug from reaching the brain. Lastly, crystal structures of α-fentanyl mAb from this study (Rodarte et al, under review) can support structure-guided design to increase affinity for fentanyl, engineer cross-reactivity to fentanyl analogs, replace potentially immunogenic amino acids, and improve protein stability to support high-concentration formulations.
Overall, the lead α-fentanyl mAbs HY6-F9_Hu (NQ) and HY11-7E1_Hu (DE) developed in this study exhibit promising in vivo efficacy and demonstrate biophysical characteristics suitable for further clinical development. These mAbs offer a promising new treatment for counteracting the effects of synthetic opioid-induced toxicity by providing an alternative to treatment with naloxone, or via dual administration of mAb and naloxone.Citation40 Further clinical development of the lead α-fentanyl mAbs described herein will grant a first-in-class opportunity to assess the clinical potential of a mAb-based therapeutic at preventing the loss of life caused by synthetic opioids.
Abbreviations
| ADA | = | anti-drug antibody |
| BLI | = | biolayer interferometry |
| CDR | = | complementarity-determining region |
| chAb | = | chimeric antibody |
| DSF | = | dynamic scanning fluorimetry |
| FDA | = | Food and Drug Administration |
| HC | = | heavy chain |
| HIC-HPLC | = | hydrophobic interaction high pressure liquid chromatography |
| LC | = | light chain |
| LCMS | = | liquid chromatography coupled mass spectroscopy |
| mAb | = | monoclonal antibody |
| MOR | = | µ-opioid receptor |
| ORF | = | open reading frame |
| OUD | = | opioid use disorder |
| PBS | = | phosphate buffered saline |
| PTM | = | post-translational modification |
| SaO2 | = | oxygen saturation |
| SEC-HPLC | = | size exclusion high pressure liquid chromatography |
| Tm | = | melting temperature |
| VH | = | heavy chain variable region |
| VL | = | light chain variable region |
Author contributions
Participated in research design: Hicks, Baehr, Pravetoni
Conducted experiments and performed data analysis: Hicks, Baehr, Silva-Ortiz, Khaimraj, Luengas, Hamid
Contributed to writing of the manuscript: Hicks, Baehr, Pravetoni
Supplemental Material
Download MS Word (250.7 KB)Acknowledgments
The authors thank Jennifer Vigliaturo from the Pravetoni Lab for LCMS technical support. The authors thank Dr. Terry L. Kirley from the University of Cincinnati College of Medicine for providing expert support in establishing the DSF method for determining Fab Tm.
Disclosure statement
Baehr, Hicks, and Pravetoni are inventors of patent applications disclosing antibodies against fentanyl and its analogs, methods of making, and use thereof. Other authors declare no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website at https://doi.org/10.1080/21645515.2022.2122507.
Additional information
Funding
References
- Ahmad FB, Rossen LM, Sutton P. Provisional drug overdose death counts; 2021.
- Centres for Disease Control and Prevention. Overdose death rates involving opioids, by type, United States, 1999-2019. National Center Injury Prev Control. 2021;2.
- O’Donnell J, Gladden RM, Goldberger BA, Mattson CL, Kariisa M. Notes from the field: opioid-involved overdose deaths with fentanyl or fentanyl analogs detected — 28 states and the District of Columbia, July 2016–december 2018. MMWR Morb Mortal Wkly Rep. 2020;69(10):1–15. doi:10.15585/mmwr.mm6910a4.
- Shover CL, Falasinnu TO, Dwyer CL, Santos NB, Cunningham NJ, Freedman RB, Vest NA, Humphreys K. Steep increases in fentanyl-related mortality west of the Mississippi River: recent evidence from county and state surveillance. Drug Alcohol Depend. 2020;216:108314. doi:10.1016/j.drugalcdep.2020.108314.
- Krotulski AJ, Chapman BP, Marks SJ, Ontiveros, ST, Devin-Holcombe, K, Fogarty, MF, Trieu, H, Logan, BK, Merchant, RC, Babu, KM. Sentanyl: a comparison of blood fentanyl concentrations and naloxone dosing after non-fatal overdose. Clin Toxicol. 2022;60(2):197–204. doi:10.1080/15563650.2021.1948558.
- Armenian P, Vo KT, Barr-Walker J, Lynch KL. Fentanyl, fentanyl analogs and novel synthetic opioids: a comprehensive review. Neuropharmacology. 2018;134:121–32. doi:10.1016/j.neuropharm.2017.10.016.
- Baumgartner JC, Radley DC. The drug overdose toll in 2020 and near-term actions for addressing it. The Commonwealth Fund; 2021.
- Martin WR. Drugs five years later: naloxone. Ann Intern Med. 1976;85(6):765. doi:10.7326/0003-4819-85-6-765.
- Rzasa Lynn R, Galinkin JL. Naloxone dosage for opioid reversal: current evidence and clinical implications. Ther Adv Drug Saf. 2018;9(1):63–88. doi:10.1177/2042098617744161.
- Ahonen J, Olkkola KT, Hynynen M, Seppälä T, Ikävalko H, Remmerie B, Salmenperä M. Comparison of alfentanil, fentanyl and sufentanil for total intravenous anaesthesia with propofol in patients undergoing coronary artery bypass surgery†. Br J Anaesth. 2000;85(4):533–40. doi:10.1093/bja/85.4.533.
- Clarke SFJ, Dargan PI, Jones AL. Naloxone in opioid poisoning: walking the tightrope. Emergency Med J. 2005;22(9):612–16. doi:10.1136/emj.2003.009613.
- Moss RB, Carlo DJ. Higher doses of naloxone are needed in the synthetic opioid era. Subst Abuse Treat Prev Policy. 2019;14(1):6. doi:10.1186/s13011-019-0195-4.
- Torralva R, Janowsky A. Noradrenergic mechanisms in Fentanyl-Mediated rapid death explain failure of naloxone in the opioid crisis. J Pharmacol Exp Ther. 2019;371(2):453–75. doi:10.1124/jpet.119.258566.
- Morell A, Terry WD, Waldmann TA. Metabolic properties of IgG subclasses in man. J Clin Invest. 1970;49(4):673–80. doi:10.1172/JCI106279.
- Mould DR, Sweeney KR. The pharmacokinetics and pharmacodynamics of monoclonal antibodies-mechanistic modeling applied to drug development. Curr Opin Drug Discovery Dev. 2007;10:84–96.
- Roiko SA, Harris AC, LeSage MG, Keyler DE, Pentel PR. Passive immunization with a nicotine-specific monoclonal antibody decreases brain nicotine levels but does not precipitate withdrawal in nicotine-dependent rats. Pharmacol Biochem Behav. 2009;93(2):105–11. doi:10.1016/j.pbb.2009.04.011.
- Raleigh MD, King SJ, Baruffaldi F, Saykao A, Hamid FA, Winston S, LeSage MG, Pentel PR, Pravetoni M. Pharmacological mechanisms underlying the efficacy of antibodies generated by a vaccine to treat oxycodone use disorder. Neuropharmacology. 2021;195:108653. doi:10.1016/j.neuropharm.2021.108653.
- Robinson C, Gradinati V, Hamid F, Baehr C, Crouse B, Averick S, Kovaliov M, Harris D, Runyon S, Baruffaldi F, et al. Therapeutic and prophylactic vaccines to counteract Fentanyl use disorders and toxicity. J Med Chem. 2020;63(23):14647–67. doi:10.1021/acs.jmedchem.0c01042.
- Baehr C, Kelcher AH, Khaimraj A, Reed DE, Pandit SG, AuCoin D, Averick S, Pravetoni M. Monoclonal antibodies counteract opioid-induced behavioral and toxic effects in mice and rats. J Pharmacol Exp Ther. 2020;375(3):469–77. doi:10.1124/jpet.120.000124.
- Ban B, Barrientos RC, Oertel T, Komla E, Whalen C, Sopko M, You Y, Banerjee P, Sulima A, Jacobson AE, et al. Novel chimeric monoclonal antibodies that block fentanyl effects and alter fentanyl biodistribution in mice. MAbs. 2021;13(1). doi:10.1080/19420862.2021.1991552.
- Smith LC, Bremer PT, Hwang CS, Zhou B, Ellis B, Hixon MS, Janda KD. Monoclonal antibodies for combating synthetic opioid intoxication. J Am Chem Soc. 2019;141(26):10489–503. doi:10.1021/jacs.9b04872.
- Wetzel HN, Webster RP, Saeed FO, Kirley TL, Ball WJ, Norman AB. Characterization of a recombinant humanized anti-cocaine monoclonal antibody produced from multiple clones for the selection of a master cell bank candidate. Biochem Biophys Res Commun. 2017;487(3):690–94. doi:10.1016/j.bbrc.2017.04.117.
- Gentry WB, Laurenzana EM, Williams DK, West JR, Berg RJ, Terlea T, Owens SM. Safety and efficiency of an anti-(+)-methamphetamine monoclonal antibody in the protection against cardiovascular and central nervous system effects of (+)-methamphetamine in rats. Int Immunopharmacol. 2006;6(6):968–77. doi:10.1016/j.intimp.2006.01.008.
- Stevens MW, Tawney RL, West CM, et al. Preclinical characterization of an anti-methamphetamine monoclonal antibody for human use. MAbs. 2014;6(2):547–55. doi:10.4161/mabs.27620.
- Stevens MW, Henry RL, Owens SM, Schutz R, Gentry WB. First human study of a chimeric anti-methamphetamine monoclonal antibody in healthy volunteers. MAbs. 2014;6(6):1649–56. doi:10.4161/19420862.2014.976431.
- Kuus-Reichel K, Grauer LS, Karavodin LM, Knott C, Krusemeier M, Kay NE. Will immunogenicity limit the use, efficacy, and future development of therapeutic monoclonal antibodies? Clin Diagn Lab Immunol. 1994;1(4):365–72. doi:10.1128/cdli.1.4.365-372.1994.
- Hwang WYK, Foote J. Immunogenicity of engineered antibodies. Methods. 2005;36(1):3–10. doi:10.1016/j.ymeth.2005.01.001.
- Harding FA, Stickler MM, Razo J, DuBridge R. The immunogenicity of humanized and fully human antibodies. Mabs. 2010;2(3):256–65. doi:10.4161/mabs.2.3.11641.
- The Antibody Society. Therapeutic monoclonal antibodies approved or in review in the European Union or the United States. 2022. http://www.antibodysociety.org/resources/approved-antibodies.
- Meyer L, López T, Espinosa R, Arias CF, Vollmers C, DuBois RM. A simplified workflow for monoclonal antibody sequencing. PLoS ONE. 2019;14(6):e0218717. doi:10.1371/journal.pone.0218717.
- Baehr C, Robinson C, Kassick A, Jahan R, Gradinati V, Averick SE, Runyon SP, Pravetoni M. Preclinical efficacy and selectivity of vaccines targeting Fentanyl, Alfentanil, Sufentanil, and Acetylfentanyl in rats. ACS Omega. 2022;7(19):16584–92. doi:10.1021/acsomega.2c00820.
- Crouse B, Wu MM, Gradinati V, Kassick AJ, Song D, Jahan R, Averick S, Runyon S, Comer SD, Pravetoni M, et al. Efficacy and selectivity of monovalent and bivalent vaccination strategies to protect against exposure to Carfentanil, Fentanyl, and their mixtures in rats. ACS Pharmacol Transl Sci. 2022;5(5):331–43. doi:10.1021/acsptsci.1c00260.
- Ehrenmann F, Lefranc M-P. Imgt/domaingapalign: IMGT standardized analysis of amino acid sequences of variable, constant, and groove domains (IG, TR, MH, IgSF, MhSF). Cold Spring Harb Protoc. 2011;2011(6):737–49. doi:10.1101/pdb.prot5636.
- Raybould MIJ, Marks C, Krawczyk K, Taddese B, Nowak J, Lewis AP, Bujotzek A, Shi J, Deane CM. Five computational developability guidelines for therapeutic antibody profiling. Proc Natl Acad Sci U S A. 2019;116(10):4025–30. doi:10.1073/pnas.1810576116.
- Swindells MB, Porter CT, Couch M, Hurst J, Abhinandan KR, Nielsen JH, Macindoe G, Hetherington J, Martin ACR. abYsis: integrated antibody sequence and structure—management, analysis, and prediction. J Mol Biol. 2017;429(3):356–64. doi:10.1016/j.jmb.2016.08.019.
- Robinson NE, Robinson AB. Deamidation of human proteins. Proc Nat Acad Sci. 2001;98(22):12409–13. doi:10.1073/pnas.221463198.
- Qiu H, Wei R, Jaworski J, Boudanova E, Hughes H, VanPatten S, Lund A, Day J, Zhou Y, McSherry T, et al. Engineering an anti-CD52 antibody for enhanced deamidation stability. Mabs. 2019;11(7):1266–75. doi:10.1080/19420862.2019.1631117.
- Cacia J, Keck R, Presta LG, Frenz J. Isomerization of an aspartic acid residue in the complementarity-determining regions of a recombinant antibody to human IgE: identification and effect on binding affinity. 1996;35(6):1897-903. doi:10.1021/bi951526c.
- Patel CN, Bauer SP, Davies J, Durbin JD, Shiyanova TL, Zhang K, Tang JX. N+1 engineering of an aspartate isomerization hotspot in the complementarity-determining region of a monoclonal antibody. J Pharm Sci. 2016;105(2):512–18. doi:10.1016/S0022-3549(15)00185-9.
- Baehr CA, Wu MM, Pandit SG, Arias-Umana J, AuCoin D, Pravetoni M. Pharmacological profiling of antifentanyl monoclonal antibodies in combination with naloxone in pre- and postexposure models of fentanyl toxicity. J Pharmacol Exp Ther. 2022;381(2):129–36. doi:10.1124/jpet.121.001048.
- Safdari Y, Farajnia S, Asgharzadeh M, Khalili M. Antibody humanization methods - a review and update. Biotechnol Genet Eng Rev. 2013;29(2):175–86. doi:10.1080/02648725.2013.801235.
- Garcês S, Demengeot J. The immunogenicity of biologic therapies. Curr Probl Dermatol. 2018;53:37–48. doi:10.1159/000478077.
- Lefranc M-P. IMGT Unique Numbering. The Immunologist. 1999;7:132–36.
- Kabat EA, Te Wu T, Perry HM, Foeller C, Gottesman KS. Sequences of proteins of immunological interest. Darby: DIANE Publishing; 1992.
- Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. J Mol Biol. 1997;273(4):927–48. doi:10.1006/jmbi.1997.1354.
- Martin AC, Cheetham JC, Rees AR. Modeling antibody hypervariable loops: a combined algorithm. Proc Natl Acad Sci U S A. 1989;86(23):9268–72. doi:10.1073/pnas.86.23.9268.
- Kunik V, Ashkenazi S, Ofran Y. Paratome: an online tool for systematic identification of antigen-binding regions in antibodies based on sequence or structure. Nucleic Acids Res. 2012;40(W1):W521–W524. doi:10.1093/nar/gks480.
- Xu Y, Wang D, Mason B, Rossomando T, Li N, Liu D, Cheung JK, Xu W, Raghava S, Katiyar A, et al. Structure, heterogeneity and developability assessment of therapeutic antibodies. MAbs. 2019;11(2):239–64. doi:10.1080/19420862.2018.1553476.
- Fekete S, Beck A, Veuthey JL, Guillarme D. Theory and practice of size exclusion chromatography for the analysis of protein aggregates. J Pharm Biomed Anal. 2014;101:161–73. doi:10.1016/j.jpba.2014.04.011.
- Haverick M, Mengisen S, Shameem M, Ambrogelly A. Separation of mAbs molecular variants by analytical hydrophobic interaction chromatography HPLC: overview and applications. Mabs. 2014;6(4):852–58. doi:10.4161/mabs.28693.
- Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen. 2001;6(6):429–40. doi:10.1177/108705710100600609.
- Lavinder JJ, Hari SB, Sullivan BJ, Magliery TJ. High-throughput thermal scanning: a general, rapid dye-binding thermal shift screen for protein engineering. J Am Chem Soc. 2009;131(11):3794–95. doi:10.1021/ja8049063.
- Ma H, Ó’fágáin C, O’Kennedy R. Antibody stability: a key to performance - analysis, influences and improvement. Biochimie. 2020;177:213–25. doi:10.1016/j.biochi.2020.08.019.
- Bailly M, Mieczkowski C, Juan V, Metwally E, Tomazela D, Baker J, Uchida M, Kofman E, Raoufi F, Motlagh S, et al. Predicting antibody developability profiles through early stage discovery screening. Mabs. 2020;12(1). doi:10.1080/19420862.2020.1743053.
- Goyon A, D’Atri V, Colas O, Fekete S, Beck A, Guillarme D. Characterization of 30 therapeutic antibodies and related products by size exclusion chromatography: feasibility assessment for future mass spectrometry hyphenation. J Chromatogr B. 2017;1065–1066:35–43. doi:10.1016/j.jchromb.2017.09.027.
- Estep P, Caffry I, Yu Y, Sun T, Cao Y, Lynaugh H, Jain T, Vásquez M, Tessier PM, Xu Y, et al. An alternative assay to hydrophobic interaction chromatography for high-throughput characterization of monoclonal antibodies. MAbs. 2015;7(3):553–61. doi:10.1080/19420862.2015.1016694.
- Wang W, Singh S, Zeng DL, King K, Nema S. Antibody structure, instability, and formulation. J Pharm Sci. 2007;96(1):1–26. doi:10.1002/jps.20727.
- Thiagarajan G, Semple A, James JK, Cheung JK, Shameem M. A comparison of biophysical characterization techniques in predicting monoclonal antibody stability. MAbs. 2016;8(6):1088–97. doi:10.1080/19420862.2016.1189048.
- Garber E, Demarest SJ. A broad range of fab stabilities within a host of therapeutic IgGs. Biochem Biophys Res Commun. 2007;355(3):751–57. doi:10.1016/j.bbrc.2007.02.042.
- Kirley TL, Norman AB, Wetzel HN. A novel differential scanning fluorimetry analysis of a humanized anti-cocaine mAb and its ligand binding characteristics. J Immunol Methods. 2020;476:112676. doi:10.1016/j.jim.2019.112676.
- Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U S A. 2017;114(5):944–49. doi:10.1073/pnas.1616408114.
- Hill R, Santhakumar R, Dewey W, Kelly E, Henderson G. Fentanyl depression of respiration: comparison with heroin and morphine. Br J Pharmacol. 2020;177(2):254–65. doi:10.1111/bph.14860.
- Pergolizzi JV, Dahan A, Ann LeQuang J, Raffa RB. Overdoses due to fentanyl and its analogues (F/FAs) push naloxone to the limit. J Clin Pharm Ther. 2021;46(6):1501–04. doi:10.1111/jcpt.13462.
- Gervais D. Protein deamidation in biopharmaceutical manufacture: understanding, control and impact. J Chem Technol Biotechnol. 2016;91(3):569–75. doi:10.1002/jctb.4850.
- Zhou K, Cao X, Bautista J, Chen Z, Hershey N, Ludwig R, Tao L, Zeng M, Das TK. Structure-function assessment and high-throughput quantification of site-specific aspartate isomerization in monoclonal antibody using a novel analytical tool kit. J Pharm Sci. 2020;109(1):422–28. doi:10.1016/j.xphs.2019.08.018.
- Yang R, Jain T, Lynaugh H, Nobrega RP, Lu X, Boland T, Burnina I, Sun T, Caffry I, Brown M, et al. Rapid assessment of oxidation via middle-down LCMS correlates with methionine side-chain solvent-accessible surface area for 121 clinical stage monoclonal antibodies. Mabs. 2017;9(4):646–53. doi:10.1080/19420862.2017.1290753.
- Lu X, Nobrega RP, Lynaugh H, Jain T, Barlow K, Boland T, Sivasubramanian A, Vásquez M, Xu Y. Deamidation and isomerization liability analysis of 131 clinical-stage antibodies. MAbs. 2019;11(1):45–57. doi:10.1080/19420862.2018.1548233.
- Nowak C, Cheung JK, Dellatore SM, Katiyar A, Bhat R, Sun J, Ponniah G, Neill A, Mason B, Beck A, et al. Forced degradation of recombinant monoclonal antibodies: a practical guide. Mabs. 2017;9(8):1217–30. doi:10.1080/19420862.2017.1368602.
- Wang SS, Yan Y, Ho K. US FDA-approved therapeutic antibodies with high-concentration formulation: summaries and perspectives. Antib Ther. 2021;4(4):262–73. doi:10.1093/abt/tbab027.
- Mathaes R, Koulov A, Joerg S, Mahler HC. Subcutaneous injection volume of biopharmaceuticals-pushing the boundaries. J Pharm Sci. 2016;105(8):2255–59. doi:10.1016/j.xphs.2016.05.029.
- Ogston-Tuck S. Intramuscular injection technique: an evidence-based approach. Nurs Stand. 2014;29(4):52–59. doi:10.7748/ns.29.4.52.e9183.
- Dear BJ, Hung JJ, Laber JR, Wilks LR, Sharma A, Truskett TM, Johnston KP. Enhancing stability and reducing viscosity of a monoclonal antibody with cosolutes by weakening protein-protein interactions. J Pharm Sci. 2019;108(8):2517–26. doi:10.1016/j.xphs.2019.03.008.
- Shahfar H, Du Q, Parupudi A, Shan L, Esfandiary R, Roberts CJ. Electrostatically driven protein–protein interactions: quantitative prediction of second osmotic virial coefficients to aid antibody design. J Phys Chem Lett. 2022;13(5):1366–72. doi:10.1021/acs.jpclett.1c03669.
- Calero-Rubio C, Saluja A, Sahin E, Roberts CJ. Predicting high-concentration interactions of monoclonal antibody solutions: comparison of theoretical approaches for strongly attractive versus repulsive conditions. J Phys Chem B. 2019;123(27):5709–20. doi:10.1021/acs.jpcb.9b03779.
- Strickley RG, Lambert WJ. A review of formulations of commercially available antibodies. J Pharm Sci. 2021;110(7):2590–608.e56. doi:10.1016/j.xphs.2021.03.017.
- Saunders KO. Conceptual approaches to modulating antibody effector functions and circulation half-life. Front Immunol. 2019;10(JUN):1296. doi:10.3389/fimmu.2019.01296.
- Hay CE, Ewing LE, Hambuchen MD, et al. The development and characterization of an scFv-Fc fusion–based gene therapy to reduce the psychostimulant effects of methamphetamine abuse. J Pharmacol Exp Ther. 2020;374(1):16–23. doi:10.1124/jpet.119.261180.
- Hedegaard H, Bastian BA, Trinidad JP, Spencer M, Warner M. Drugs most frequently involved in drug overdose deaths: United States, 2011-2016. Natl Vital Stat Rep. 2018;67:1–14.
- Lockwood TLE, Huynh P, Richard A, Sightes E, Bailey K, Ray B, Lieberman M. Community overdose surveillance: comparing substances collected from the death scene investigation to toxicology results. Drug Alcohol Depend. 2021;224:108722. doi:10.1016/j.drugalcdep.2021.108722.