
ABSTRACT
In this study, we sought to evaluate the inter-batch consistency and safety of the CTN-1 V human rabies vaccine (Vero cells). A total of 594 healthy participants aged 10–60 years were enrolled from Mianzhu, Sichuan Province, and randomized into three batch groups to receive vaccination via the Essen Regimen, that is, a single dose on days 0, 3, 7, 14, and 28 in the deltoid muscle of the upper arm. The serum antibody geometric mean concentration (GMC) and positive conversion rate of each group were determined using a rapid fluorescence focus inhibition test (RFFIT) before the first-dose immunization, 14 d after the first-dose immunization, and 14 d and 12 mo after full immunization. Adverse events (AEs) 30 min and 30 d after immunization were observed in each group. There were 322 cases of AEs during the observation period, with an overall incidence of 54.4%. The incidences of AEs in groups A, B, and C were 57.4%, 51.5%, and 54.3%, respectively. There were no significant differences among the groups (P > .05). Moreover, there were no significant differences (P > .05) in the serum GMC or antibody-positive conversion rate between any two groups at any time point. The bilateral 95% confidence interval of the GMC ratio between any two groups 14 d after the first-dose immunization was within the range of 0.67–1.50. This study shows that the CTN-1 V human rabies vaccine (Vero cells) has reliable safety and stable immunogenicity between batches.
Plain Language Summary
This was a randomized, double-blind, equivalent clinical study on the inter-batch consistency of rabies vaccine. The rabies virus CTN strain adopted for the vaccine was isolated by China National Institute for Food and Drug Control. It has a gene sequence homology of 82.0%–93.0% with the representative strains of street virus isolated in China (i.e., CQ92, HN06, and J strains), higher than that of other vaccine strains (aG, PM, and PV strains). A total of 594 participants were enrolled in this study, and were randomized into three batch groups. Blood samples were collected from participants in each group before vaccination, at 14 days after the first dose of vaccination, and at 14 d and 12 mo after the fifth dose of vaccination, in order to detect antibody levels and observe adverse reactions. There was no significant difference in serum antibody levels and adverse reactions between any two of the three groups, indicating that different vaccine batches manufactured at the commercial scale can maintain stable immunogenicity and safety profiles.
Introduction
Rabies is a widespread and dangerous zoonotic disease caused by infection with the rabies virus. Rabies epidemics are estimated to have affected more than 150 countries, causing approximately 59,000 deaths per year, with 95.0% of cases in Africa and Asia.Citation1,Citation2 When the rabies virus reaches the central nervous system of the human body, it is fatal.Citation3 Pre-exposure and timely post-exposure vaccinations are the most effective methods to prevent rabies.Citation4
The CTN-1 V strain isolated by the China National Institute for Food and Drug Control has been adopted as the production strain for the human rabies vaccine manufactured by Dalian Aleph Biomedical Co., Ltd., because the gene sequence of the CTN-1 V strain is more homologous to that of the current street-virus epidemic strain, and shows good immunogenicity.Citation5,Citation6 In the randomized, double-blind, and positive-controlled clinical trials of the vaccine before marketing, the results showed that, at 14 d after the first dose of vaccination and at 14 d after full vaccination, the antibody positive conversion rates were 100%, and the GMCs, were 9.96 and 28.83 IU/mL, respectively; the local reactions were mainly pain, redness, swelling, and itching at the inoculation site, and the systemic reactions were mainly fever, headache, and fatigue, most of which were mild (Grade 1) in severity.Citation7
This study was conducted to evaluate equivalence between any two of three vaccine batches manufactured consecutively at the commercial scale. The inter-batch consistency could be confirmed only when equivalence assumption was deemed valid between any two of the three batches at the same time, and the influence of confounding factors, such as participants’ antibody levels before immunization, study site effect, and evaluation time point after immunization, must be strictly controlled, in order to ensure the validity of the evaluation.
For participants with positive antibody results before immunization, re-vaccination is similar to booster immunization, while the effect of booster immunization may vary greatly among participants due to differences in immunization history; for a population exposed to Class III rabies, the principle of post-exposure treatment requires rabies-specific immunoglobulin. Both these aspects are key points to be considered in determining the subject population for primary endpoint evaluation and are confounding factors that seriously affect the inter-batch consistency evaluation. Therefore, the main evaluation population comprised participants who had no immunization history, had negative antibody results before immunization, and were currently free from rabies exposure.
A single-center design was adopted in this clinical study to exclude the influence of study site effect on inter-batch consistency evaluation, and a city with a low vaccination rate of rabies vaccine (Mianzhu City) in a western province of China (Sichuan) was selected as the study site.
For evaluation time point, a longer interval from the first immunization might result in more factors affecting the antibody levels; thus, the evaluation time point should be as early as possible. Besides, an antibody positive conversion rate of 100% (close to the antibody peak level) could be achieved at 14 d after the first immunization via Essen Regimen; therefore, we determined that the best time point for equivalence verification was 14 d. Furthermore, the difference test was planned to be carried out at 14 d and 12 mo after full immunization.
Considering the sensitivity of the evaluation, the GMC of the neutralizing antibody was selected as the main evaluation indicator, instead of antibody positive conversion rate; the latter could only be used to evaluate whether the serum antibody level reached a protective level (≥ 0.5 IU/mL), but cannot be used for inter-batch difference analysis.
Materials and methods
Study participants
A total of 592 healthy volunteers were screened at the Center for Disease Control and Prevention of Mianzhu, Sichuan Province, China, between May 19, 2019 and August 6, 2020. The volunteers were randomized into three groups (Batch groups 1, 2, and 3) to receive vaccination via the Essen Regimen. The healthy volunteers were recruited by publishing recruitment advertisements at local public places and media. The volunteers visited the CDC and participated in this study voluntarily. All study participants provided informed consent. For juveniles aged 10–18 years, we provided a specific informed consent form (juvenile version) and explained the contents to them, so that they can be informed independently. In addition, their guardians were subjected to an informed consent process using an adult version of the informed consent form; contents (including all intervention procedures in the protocol) in the ICF versions for both juveniles and adults comply with the ICF GCP.
The main criteria for inclusion were as follows: aged 10–60 years, voluntarily participating in the study and signing informed consent form; no history of receiving rabies vaccine or rabies virus passive immunization agents; no history of allergy to or serious adverse effects from any vaccine component or drug; no serious history of immunodeficiency disease and heart disease that interfere with the conduct of the study; females of childbearing age who have negative urine pregnancy test results and have no fertility plan during the study (for details, please refer to ”China Registration and Information Publicity Platform for Drug Trials” http://www.chinadrugtrials.org.cn/index.html, with the registration number: CTR20191083).
The main exclusion criteria comprised the exclusion criteria for both first dose and immune persistence observation and can be summarized as follows. 1) Exclusion criteria for the first dose: (1) history of a bite from a rabies-susceptible animal (e.g., cats and dogs) within the past year or receiving either the rabies vaccine or passive immunization agents; (2) acute disease or an acute attack of chronic disease within the past 3 d; (3) receiving any vaccine within the past 14 d; (4) using other investigational or unregistered products (drugs or vaccines) within the past month or having plans to participate in other clinical studies within 7 mo of enrolling in this study (within 13 mo of enrollment for the immune persistence observation subgroup); (5) receiving or having received blood transfusion, blood products, immunoglobulins, or other immune enhancers within the past 4 mo; (6) history of severe allergy to any of the components of the study vaccine (sodium chloride, potassium chloride, potassium dihydrogen phosphate, disodium hydrogen phosphate, human albumin, and thiomersal) or antibiotics that might induce adverse effects such as anaphylactic shock, allergic laryngeal edema, allergic purpura, thrombocytopenic purpura, or local hypersensitive necrosis reaction (Arthus reaction); (7) history of serious vaccine- or drug-induced adverse effects, including allergy, urticaria, skin eczema, dyspnea, or angioneuroedema; (8) congenital or acquired immunodeficiency or receiving immunosuppressive treatment within the past 3 mo, such as long-term systemic corticosteroid therapy (prednisone or similar drugs that have been administered for more than 2 consecutive weeks); (9) severe congenital malformations or chronic diseases that may have interfered with the conduct or completion of the study (including but not limited to Down syndrome, thalassemia, heart or kidney disease, diabetes, uncontrolled hypertension (blood pressure higher than 120/80 mmHg for participants aged 10–12 years and 140/90 mmHg for participants aged 13–60 years), autoimmune diseases, hereditary allergic constitution, or Guillain-Barré syndrome; (10) systemic diseases with clinical or serological evidence, such as tuberculosis, hepatitis B, hepatitis C, or human immunodeficiency virus (HIV) infections; (11) history or family history of convulsions, epilepsy, encephalopathy, or psychosis; (12) contraindications to intramuscular injection, such as thrombocytopenia, the presence of a coagulation disorder, or anticoagulant treatment; (13) splenectomy or participants with a dysfunctional spleen; (14) females who were pregnant or lactating, or female participants of childbearing age with a positive urine pregnancy test, or those planning to get pregnant within 2 mo; (15) participants planning to move before the end of the study or leaving the local area for a long time during the scheduled study visit; and (16) any situation that the investigator believed may interfere with trial evaluation. 2) Exclusion criteria for immune persistence observation were as follows: (1) the top 210 participants who had not completed whole-course vaccination; (2) participants from whom the blood samples had not been collected before the first vaccination or 14 d after whole-course vaccination; (3) participants who were administered the rabies immunoglobulin or any other rabies vaccine within 12 mo of whole-course vaccination; (4) participants, who after vaccination, received long-term (≥15 d) immunosuppressant therapy or other immunomodulatory drugs (such as glucocorticosteroids) that may affect the immune response of the rabies vaccine; and (5) any situation that the investigator believed may interfere with trial evaluation.
The main objective of this study was to verify the consistency of immunogenicity among the three consecutive commercial-scale batches. As the inter-batch consistency evaluation requires equivalence among the three batches, a healthy population with smaller variability and GMC with higher sensitivity were selected as evaluation indicators.
The study strictly followed the Good Clinical Practice and the Declaration of Helsinki and was approved by the Vaccine Clinical Trial Ethics Committee of the Sichuan Provincial Center for Disease Control and Prevention (SC-0820190201). There was no change in test methods or indexes during the study.
Vaccines
The vaccines used in this study were prepared by Dalian Aleph Biomedical Co., Ltd. (strength: 1.0 mL/vial; dose: 1.0 mL/vial for each human dose; dosage form: liquid vaccine). The vaccines were transported and stored at 2–8°C. The antigen titer of Batch group 1 (batch No.: 201810025, expiry date: April 27, 2020) is 7.6 IU/dose, Batch group 2 (batch No.: 201810026, expiry date: April 27, 2020) is 7.5 IU/dose, and Batch group 3 (batch No.: 201811027, expiry date: May 5, 2020) is 8.4 IU/dose.
Vaccination
The participants in three batch groups were inoculated with one dose of the test vaccine intramuscularly in the deltoid muscle of the upper arm using the Essen Regimen on days 0, 3, 7, 14, and 28. The vaccines were fully shaken before use.
Safety observation
Solicited and unsolicited AEs from the first dose to 30 d after full vaccination, and all serious adverse events (SAEs) from the first dose to 6 mo after full vaccination were recorded. Adverse events were graded and evaluated in accordance with the Guidelines for Grading Criteria for Clinical Adverse Reactions to Vaccines for Prophylaxis published by National Medical Products Administration (NMPA).Citation8
Immunogenicity observation
Five milliliters of venous blood was collected from all participants before the first vaccination, 14 d after the first vaccination, and 14 d and 12 mo after full vaccination to separate serum, which was stored at −20°C. Serum GMC and neutralizing antibody-positive conversion rate before and after immunization were determined by the National Institutes for Food and Drug Control using the rapid fluorescent focus inhibition test (RFFIT).Citation9
The RFFIT is the standard, WHO-recommended method for detecting rabies virus neutralizing antibodies (RVNAs). RVNAs in serum samples were neutralized with the CVS strain of the rabies virus as the challenge virus. A suspension of susceptible cells containing the viral residue was used, and the virus was detected using fluorescent antibody staining. A fixed amount of the CVS virus was incubated with serially diluted serum samples (in vitro neutralization reaction) and was then added to the suspension of susceptible cells. After 24-h incubation, the cells were fixed with acetone in a monolayer and stained with fluorescence-labeled anti-NP antibodies to detect the unneutralized virus. Compared to the virus control group, the dilution amount required for reducing the concentration of a fixed amount of CVS virus (FFU/mL) by 50% was calculated, and the neutralizing antibody titer in each serum sample was determined by comparing against a reference serum with a known titer.
Positive conversion criteria were as follows: serum neutralizing antibody <0.5 IU/mL before immunization and ≥0.5 IU/mL after immunization.Citation10,Citation11 Negative conversion criterion was as follows: serum neutralizing antibody <0.5 IU/mL after immunization.
Between-run consistency analysis
According to the WHO’s “Guidelines on the clinical evaluation of vaccines: regulatory expectations, Annex 9, TRS No 1004,” it is suggested to demonstrate non-inferiority based on GMT, and the lower bound of the 95% confidence interval (CI) should not fall below 0.67 or 0.5 (polyvalent vaccine). An equivalence study on inter-batch consistency requires both lower and upper limits to be within the equivalence margin. Referring to the standard of non-inferiority, the upper limit was set to 1.5; thus, the equivalence margin was set to 0.67–1.5.
The evaluation criteria for equivalency testing: 14 d after the first dose, the 95% CI of the GMC ratio between any two batches was between 0.67 and 1.50.
Sample sizes
It was assumed that the pairwise GMC ratios between any two of the three batches were equivalent after full vaccination. The standard deviation after logarithmic transformation (with the logarithm base of 10) was 0.45, equivalence margin after logarithmic transformation (with the logarithm base of 10) was 0.176, test significance level was 0.05 (two-sided), overall power was 80%, and power of each hypothesis test was 93.3%, Bonferroni method was used for adjustment. The sample size required for each group was calculated using PASS software, and it was determined to be 159 participants. After a comprehensive consideration of a pre-immune positive rate of 5.0% and a dropout rate of 15.0%, 198 participants were planned to be enrolled in each group, with 594 participants enrolled in total. The first 210 participants enrolled were observed for immunopersistence.
The calculation of the sample size using PASS (Power Analysis and Sample Size) software is as follows:
1) Open PASS 15.0.5 software and enter in sequence Means/Two Independent Means/Efficiency/Efficiency Tests for the difference Between Two Means (Test Version).
2) Calculation Program:
For more details, please refer to .
Figure 1. Calculation program.

3) Result:
For more details, please refer to .
Table 1. Testing equivalence of two means using a parallel-group design.
4) Summary Statements
An equivalence test of means performed using two one-sided tests on data from a parallel-group design with sample sizes of 159 in the reference group and 159 in the treatment group achieves 93% power at a 5.0% significance level when the true difference between the means is 0.0, standard deviation is 0.5, and equivalence limits are −0.2 and 0.2.
For more details, please refer to .
Table 2. Dropout-Inflated sample size.
Randomization method
Randomization was performed using a ratio of 1:1:1 according to the randomized block method. The random allocation table was generated by a randomization statistician using SAS statistical software (version 9.4). Study No. Range: 0001–0594.
Blinding
A double-blind design was adopted for this study. Vaccine blinding was performed by the randomization statistician and pharmacists. That is, a printed label was affixed to the designated position of each vaccine according to the blind codes. Vaccine blinding was supervised by the randomization statistician, who guided blinding personnel to conduct labeling according to the blind codes. The blind codes were sealed up for safekeeping after blinding. The entire blinding process was documented. The blinding personnel were not allowed to participate in other work related to the clinical study or disclose the blind codes to any person involved in the clinical study. The three batches of vaccines involved in this clinical study were consistent in appearance and package, and therefore, the data were be collected in a double-blind manner. During the study, grouping information was kept confidential from all participants (including their parents or other legal guardians), investigators, and project members involved in any endpoint evaluation, data review, or data analysis of the study.
Statistical analysis
All statistical analyses were processed using SAS software (version 9.4), and all measurement data are presented as X ± SD, with the difference test performed using variance analysis. All count data are expressed by either frequency number or frequency and composition ratio, with the difference test performed using the chi-square test or Fisher’s exact probability method. The two-sided significance level (α) of 0.05 was adopted. The 95% CI of the antibody-positive conversion rate was calculated using the Clopper–Pearson method. The antibody GMCs of the pre-immune antibody-negative population 14 d after the first-dose immunization were subject to logarithmic transformation, and the analysis of covariance (ANCOVA) model was fitted for equivalence analysis. Antibody GMCs at the remaining time points were subject to logarithmic transformation in order to statistically analyze inter-group differences.
Criteria for suspension or termination of the study
The occurrence of vaccine-related death or life-threatening SAEs in any subject during the study was determined as follows: occurrence of AEs of grade ≥ 3 during the 30-d follow-up period after each dose in ≥15% of people vaccinated.
Results
Subject recruitment for this study began on May 17, 2019, and ended on August 2, 2019. Follow-up lasted until August 6, 2020, 12 mo after full immunization in the last subject. A total of 594 participants were randomized into three batch groups, each with 198 participants; 187, 182, and 189 participants completed all five doses of vaccination, respectively. A total of 37 participants dropped out during the study (i.e., 11, 16, and 10, respectively), mainly because they were busy with farming. In the three groups, 197, 198, and 197 participants, respectively were included in the safety analysis set (SS, participants receiving at least 1 dose of vaccination); 186, 184, and 189 participants, respectively were included in the per protocol set at 14 d after the first dose of vaccination (PPS1, which referred to the dataset of participants meeting the blood sample collection criteria at 14 d after the first dose of vaccination). Each subject met all of the following conditions: the inclusion criteria, did not meet the exclusion criteria, underwent randomization, completed the first dose of vaccination, completed immunogenicity blood collection pre-dose and 14 d after the first dose of vaccination, and had valid antibody level results). In total, 186, 181, and 186 participants, respectively were included in per protocol set at 14 d after full vaccination (PPS2, which referred to the dataset of participants meeting the blood sample collection criteria at 14 d after full vaccination). Each subject met all of the following conditions: the inclusion criteria, did not meet the exclusion criteria, underwent randomization, completed the first dose of vaccination, completed immunogenicity blood collection pre-dose and 14 d after full vaccination, and had valid antibody level results.
Please refer to and for more details.
Figure 2. Subject disposition flowchart.
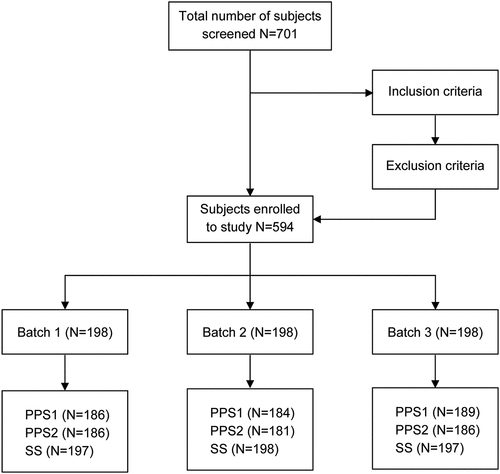
Table 3. List of participants who dropped out.
Baseline characteristics of the participants
All 592 participants completed the enrollment. There were no significant differences in the demographics and other baseline characteristics among the groups (P > .05).
Please refer to for more details.
Table 4. Baseline demographic characteristics of participants (SS).
Safety
Incidence of adverse events
In this study, a total of 592 participants completed vaccination and safety monitoring. During the observation period, 322 participants showed AEs. The overall incidence of AEs was 54.4%; there were 113 (57.4%), 102 (51.5%), and 107 (54.3%) participants with AEs who received vaccines of batches 1 through 3, respectively. There were no significant differences among the groups (P > .05). The severity of the AEs was mainly mild (grade 1). A total of 226 participants showed local AEs, with an overall incidence of 38.2%; the incidence of AEs in the three groups was 40.1%, 34.3%, and 40.1%, with predominant Grade 1 pain (31.8%), redness (12.3%), swelling (7.8%), pruritus (3.9%), induration (2.0%), and rash (0.2%). A total of 148 participants showed systemic AEs, with an overall incidence of 25.0%; the incidence of systemic AEs in the three groups was 22.3%, 26.8%, and 25.9%, with predominant Grade 1 Pyrexia (15.9%). There were no significant differences in the symptoms and severity of local and systemic reactions among the groups (P > .05). The occurrence of AEs, local and systemic, in each group is shown in .
Table 5. Incidence of AEs in different groups (SS).
Table 6. Severity of adverse effects (AEs) (SS).
Nine SAEs occurred during the observation period. All SAEs were due to hospitalization, and the outcomes were all symptom resolution (without sequelae), with no relationship with the vaccination.
Pregnancy events
A total of three pregnancy events occurred during the study. One subject was confirmed pregnant after four doses of the vaccine, and one subject was confirmed pregnant after five doses of the vaccine. Both participants delivered successfully, and the babies were normal at the visit 1 mo after delivery. One subject was confirmed pregnant after five doses of the vaccine and chose to terminate the pregnancy voluntarily. No pregnancy-related SAEs occurred.
Immunogenicity
In the three batch groups, the proportions of participants with GMC < 0.5 IU/mL before immunity were 94.4% (186/197), 93.9% (186/198), and 91.4% (180/197), for groups 1 through 3. Of the 524 participants who completed the first immunization dose, 519 completed the full vaccination and 182 completed the immunogenicity observation 12 mo after the full vaccination.
In the three batch groups, the antibody-positive conversion rate was 100.0% 14 d after the first dose of immunization and 14 d after the full vaccination; the antibody-positive conversion rates were 77.8%, 84.8%, and 91.7% 12 mo after the full vaccination, for groups 1 through 3, respectively. The antibody GMC was 28.54, 29.76, and 34.99 IU/mL 14 d after the first dose of immunization, 21.34, 24.90, and 27.38 IU/mL 14 d after the full vaccination; and 1.34, 1.44, and 1.48 IU/mL 12 mo after the full vaccination, for groups 1 through 3, respectively. As shown in , there were no significant differences in the antibody-positive conversion rate and GMC among the three groups of participants vaccinated at different times (P > .05).
Table 7. Serum neutralizing antibody-positive conversion rate and GMC at different observation time points in the pre-immune negative population (PPS1/PPS2).
Inter-batch consistency
First, as the immunogenic reaction and antibody maintenance ability differ between participants, the seroconversion rate in different groups at different test points was expected to vary. Usually, seroconversion reaches 100% at 14 d after full-course vaccination. The earlier the time before the full vaccination, the greater the difference between the groups. Second, the potency differed among vaccines from different batches (see 1.2 Vaccines).
As shown in , the bilateral 95% CI of the GMC ratio in both groups ranged from 0.67 to 1.50 at 14 d after the first dose of immunization.
Table 8. Validation of antibody GMC equivalence 14 d after the first dose of immunization (PPS1).
Discussion
Data from some clinical studies have shown that the immune effect of purified Vero cell rabies vaccine is similar to the protective effect and safety of HDCV, with a low price and large yield, making it one of the rabies vaccines recommended by the World Health Organization. It has been widely promoted and applied around the world.Citation12,Citation13
The results of this study showed that the overall incidence of adverse reactions was 50.5%, incidence of local reactions was 38.2%, and incidence of systemic reactions was 25.0%; the common symptoms after vaccination mainly included Grade 1 (mild) pain, pyrexia, redness, and swelling, similar to those reported by Jie et al.Citation14 (54.5%); the incidences of adverse reactions were comparable among the three batches.
Detection of rabies virus neutralization antibody(RVNA) is considered the simplest method for rabies exposure immune reaction; RVNA ≥ IU/mL indicates that the organism has protective function against virus exposure. The results of this study showed that the antibody positive conversion rate was 100% at 14 d after the first dose and 14 d after full vaccination, and the GMC was higher than 21.34 IU/mL; the antibody positive conversion rate remained at 77.8%–91.7% at 12 mo after vaccination, and the antibody GMC remained at 1.34–1.48 IU/mL, indicating that the investigational vaccine has favorable immunogenicity and immunopersistence. The results of this study are similar to those reported by Xinxiong et al.Citation15 and Lirong et al.Citation16 The GMC interval equivalence evaluation 14 d after the first dose showed that the bilateral 95% CI of GMC ratio between any two of the three groups ranged from 0.67 to 1.50, and the inter-batch equivalence was verified.
The results of this clinical study showed that three consecutive batches of CTN-1 V strain human rabies vaccine (Vero cells) had reliable safety and stable immunogenicity, and the inter-batch consistency met the requirements.
Trial registration
CTR20191083
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The protocol is saved at Dalian Aleph Biomedical Co., Ltd. and is available from the corresponding authors if necessary.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Hm F Jr, Petersen BW, Robertson KL, Rupprecht CE. Rabies: still a uniformly fatal disease? Historical occurrence, epidemiological trends, and paradigm shifts. Curr Infect Dis Rep. 2012;14(4):1–9. doi:10.1007/s11908-012-0268-2. PMID: 22699971.
- Hampson K, Coudeville L, Lembo T, Sambo M, Kieffer A, Attlan M, Barrat J, Blanton JD, Briggs DJ, Cleaveland S, et al. Estimating the global burden of endemic canine rabies [published correction appears in PLoS Negl Trop Dis. 2015 May;9(5):e0003786]. PLoS Negl Trop Dis. 2015;9(4):e0003709. doi:10.1371/journal.pntd.0003709. PMID: 25881058.
- Pichon S, Guinet-Morlot F, Minutello M, Donazzolo Y, Rouzier R, Chassard D, Fitoussi S, Hou V. A serum-free, purified Vero cell rabies vaccine is safe and as immunogenic as the reference vaccine Verorab for pre-exposure use in healthy adults: results from a randomized controlled phase-II trial. Vaccine. 2013;31(18):2295–301. doi:10.1016/j.vaccine.2013.02.058. PMID: 23510665
- World Health Organization. WHO expert consultation on rabies: third report. WHO; 2008. https://apps.who.int/iris/handle/10665/272364.
- Guanmu D, Gelin X, Qiyou X, Dingming W, Yuemei H, Dunjin Z, Ping W, Yongzhen Z, Xiaoming Y, Fengcai Z, et al. Epidemiological investigation of rabies virus in domestic dogs, cats, and wild animals and observation on the immune effect of different rabies vaccines in China. Chin J Virol. 2007;23(6):417–23. doi:10.3321/j.1000-8721.2007.06.001.
- Manrong C, Jie W, Shengli M, Gelin X. Protection of CTN-1V human rabies vaccine prepared by CTN strain against challenge with representative rabies street virus strains in China. Chin J Biol. 2008;21(9):786–87. doi:10.13200/j.cjb.2008.09.55.chengmr.018.
- Huang T, Li Y, Mo S, Li J, Nan R, Zhou L, Wang M, Nie F, Chen J, Yang T, et al. Safety and immunogenicity of domestic rabies vaccine (Vero cells) for human use prepared with CTN-1V strain in healthy population. Chin J Biologicals. 2018;31(6):624–628, 637. doi:10.13200/j.cnki.cjb.002201.
- National Medical Products Administration. About printing and distributing six technical guidelines including ”technical guidelines for preclinical study of preventive vaccines”. NMPA; 2005. https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjyp/20051014010101369_5.html.
- Ling Z, Tianxi Z, Kewen S, Siwei T, Ji S, Haipeng Y. Evaluation of the efficacy of rapid fluorescent foci inhibition test for the detection of rabies virus neutralizing antibodies. Chin J Health Inspection. 2021;31:2076–79.
- World Health Organization. Rabies vaccines: WHO position paper, April 2018 - Recommendations. Vaccine. 2018;36(37):5500–03. doi:10.1016/j.vaccine.2018.06.061. PMID: 30107991.
- Fishbein DB, Yenne KM, Dreesen DW, Teplis CF, Mehta N, Briggs DJ. Risk factors for systemic hypersensitivity reactions after booster vaccinations with human diploid cell rabies vaccine: a nationwide prospective study. Vaccine. 1993;11(14):1390–94. doi:10.1016/0264-410x(93)90167-v. PMID: 8310759
- Yanrong L. Analysis of epidemic characteristics of Chinese rabies from 2007 to 2016 and forecasting of epidemic trends. Jiamusi University; 2018. https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CMFD&dbname=CMFD201901&filename=1018876800.nh&uniplatform=NZKPT&v=HPkAOMK-ggFkcTUTzG5227O0CuX7sWxdmYjroS-nP0UCuMNeNrmD45zrKspee1E5.
- Haupt W. Rabies-risk of exposure and current trends in prevention of human cases. Vaccine. 1999;17(13–14):1742–49. doi:10.1016/s0264-410x(98)00447-2. PMID: 10194833
- Manning SE, Rupprecht CE, Fishbein D, Hanlon CA, Lumlertdacha B, Guerra M, Meltzer MI, Dhankhar P, Vaidya SA, Jenkins SR, et al. Human rabies prevention-United States, 2008: recommendations of the advisory committee on immunization practices. MMWR Recomm Rep. 2008;57(RR–3):1–28. PMID: 18496505.
- Xinxiong Z, Yutao Z, Wei C, Wen S, Huafang D, Shi H, Zuolin H, Lingbing Z, Gelin X. Clinical trials of adjuvant-free freeze-dried rabies vaccine for human use. Chin J Biol. 2007;4:276–77. doi:10.13200/j.cjb.2007.04.49.zhengxx.014.
- Lirong H, Tengrong L, Guiqiu P, Shouchun C, Wei Z, Yuting H, Mingli L, Yanping L. Safety and immunogenicity of freeze-dried rabies vaccine (Vero cells) for human use in healthy population at ages of 10 - 60 years. Chin J Biol. 2017;30(3):283–87. doi:10.13200/j.cnki.cjb.001685.