
ABSTRACT
Cancer-testis antigen CT23 is a class of tumor-associated antigens (TAA) characterized by restricted expression in male germ cells and a variety of tumor tissues. Numerous studies have shown that CT23 is closely related to tumor cell viability, proliferation, metastasis and invasion. CT23 is immunogenic and can cause specific immune response in tumor patients. Therefore, it is considered to be one of the best target antigens for designing therapeutic tumor vaccines and T-cell-mediated tumor immunotherapy. In this study, we initially obtained seven HLA-A*0201-restricted CT23 epitope candidate peptides through the T cell epitope prediction program. Subsequently, a T2 cell binding assay revealed the potential binding of all candidate peptides with HLA-A2 molecules. Notably, peptide P7 (ALLVLCYSI) exhibited the highest affinity, as evidenced by a fluorescence index (FI) of 2.19. Dendritic cells (DCs) loaded with CT23 candidate peptide can stimulate CD8+T cell activation and proliferation, and compared with other candidate peptides, candidate peptide P7 is superior. The cytotoxic T lymphocytes (CTLs) stimulated by the peptide P7 had killing effect on tumor cells (HLA-A*0201+, CT23+), but no killing effect on tumor cells (HLA-A*0201−, CT23+). The CTLs induced by the peptide P7 also had a specific killing effect on T2 cells bearing the peptide P7. In summary, our findings suggest that the CT23 peptide P7 (ALLVLCYSI) can induce immune responses and holds potential for tumor-specific CTL therapy.
Introduction
Cancer-testis antigen (CTA) is a class of tumor-associated antigens (TAA) characterized by restricted expression in male germ cells and a variety of tumor tissues, while it is rarely expressed in normal adult tissues.Citation1 At present, the CTA public database (www.cta.lncc.br) has included more than 200 CTA gene families. There are divided into two categories based on their chromosomal location: (1) CTX, coding genes located on the X chromosome, including MAGE, GAGE and NY-ESO-1; (2) Non-CTX, encoding genes to be mapped on non-X chromosomes, such as CT23, SCP-1 and AKAP3/4. Numerous studies have demonstrated that many CTAs are closely related to tumor cell viability, proliferation, metastasis and invasion.Citation2,Citation3 Testes are immune-exempt organs due to the presence of blood-testis barrier and the absence of human leukocyte antigen (HLA) molecules expressed by germ cells. The presence of high levels of CTA in tumor tissues enables the immune system to recognize tumor cells as ‘non-self,’ thereby triggering an anti-tumor immune response. Importantly, the minimal expression of CTA in adult normal tissues makes it a promising target antigen for the development of therapeutic vaccines and for T-cell-mediated tumor immunotherapy by oncologists.Citation3–6 Among identified CTAs, NY-ESO-1 and MAGEs are the two most extensively studied and widely used CTAs in the treatment of malignant tumors. NY-ESO-1 accounts for 37% of all clinical CTA trials, while MAGEs account for 36%. These antigens have shown the ability to stimulate cellular and humoral immune responses, demonstrating both safety and antigenicity.Citation5,Citation6 For instance, a HLA-A2-binding NY-ESO-1 peptide vaccine generated a detectable specific immune response in 75% (9/12) of patients with NY-ESO-1-expressing cancers.Citation7 Moreover, a previous clinical trial targeting MAGE-3 reported significant tumor regression in 28% of patients with melanoma (7/25) who received peptide vaccine treatment.Citation8 In addition, vaccines targeting other CTAs, such as LY6K (also known as URLC10), TTK1, IMP-3, PRAME, and SP17, have been put into several clinical trials and have shown to control tumor progression.Citation3,Citation6 Although CTA-targeted therapy has always received much attention, only a few of CTA targets have been tested for efficacy in clinical trials.
Acrosin binding protein (ACRBP), also known as OY-TES-1, was identified and named by Ono et al in 2001, and was also called CT23 because it ranked 23rd in the CTA family. CT23 gene is located on human chromosome 12p12 ~ 13, which has a total length of 9339 bp, contains 10 exons, and a full-length transcription unit of 1895 bp.Citation9 Previous studies have confirmed that CT23 is highly expressed in various tumor tissues.Citation9–17 It was found that CT23 could promote cancer cell proliferation by inhibiting NuMA (Nuclear Mitotic Apparatus Protein) to normalize mitotic spindle function.Citation10 Some of experiments in vitro have confirmed that down-regulating CT23 can reduce cancer cell growth, prevent cancer cell migration and invasion, and elevate the expression of apoptosis-related proteins, thereby promoting cell apoptosis.Citation11,Citation12 In patients with ovarian cancer, high expression of CT23 was associated with paclitaxel resistance, stage and chemotherapy sensitivity, shortened survival time and accelerated recurrence rate,Citation10,Citation13 while in colorectal cancer patients, high expression of CT23 is also confirmed to be correlated with tumor invasion stage and histological grade.Citation15 Furthermore, the presence of CT23 antibodies in the serum of patients with various tumors, including ovarian cancer, colorectal cancer, liver cancer, and glioma, indicates that CT23 is immunogenic and can induce a specific immune response,Citation13–15,Citation17 making it a potential diagnostic and therapeutic target in cancer.
Tumor antigens are processed into peptides in cells, and presented to the surface of tumor cells by HLA class I molecules. CD8+T lymphocytes can be activated after specific recognition of tumor antigen peptides, and differentiate into CTLs that can specifically kill tumor cells. CTLs are considered to be the most crucial effector cell in body anti-tumor immunity. Therefore, therapeutic tumor vaccine based on HLA class I molecular restriction epitopes is one of the vital strategies for integrative biological therapy of tumors.Citation18 Given the high expression and immunogenicity of CT23 in a variety of tumor tissues, and the prevalence of HLA-A2, particularly its subtype HLA-A*0201, in the Chinese population,Citation19 the identification of CT23 epitopes presented by HLA-A*0201 molecule and its application in tumor immunotherapy holds certain clinical significance. In this study, several public databases were accessed to predict HLA-A*0201 restricted peptides from CT23 protein sequences. Next, binding affinity and stability of CT23 candidate peptides with HLA-A2 molecule were analyzed by the T2 cell binding assay, and DCs were sensitized with CT23 candidate peptides in vitro to induce specific CTL generation. The dominant epitopes of CT23 were screened through enzyme linked immunospot assay and cytotoxicity assay, laying a foundation for further exploration of tumor immunotherapy based on CT23 peptides.
Material and methods
Cell lines and human peripheral blood samples
Glioma cell line (U87), and ovarian cancer cell lines (OVCAR-3, SKOV3) were obtained from the Shanghai Cell Collection of the Chinese Academy of Science. T2 cell was generously provided by professor Xiao-ling Lu (Guangxi Medical University, Nanning, Guangxi, P. R. China). All of cell lines were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS) at 37°C in an atmosphere containing 5% CO2.
The peripheral blood samples were collected from HLA-A2+ healthy donors. All experiments were conducted following the guidelines and regulations of the Ethical Review Committee of Guangxi Medical University and informed consent was obtained from all donors.
Recombinant protein generation
The sequence of CT23 open reading framework was amplified by PCR, and the PCR products were inserted into plasmid pMAL-c2 to construct CT23 prokaryotic expression plasmid and induce the expression of recombinant CT23 protein. The specific steps were described in our previous report.Citation15
Epitope peptide prediction and synthesis
According to the binding ability of antigen peptides to HLA-I molecules, C-terminal enzyme restriction site in proteasome and transporter-associated antigen processing (TAP) transport efficiency, computational methods for predicting CD8+T cell epitopes have been well developed and increasingly mature, with high speed and low cost characteristics.Citation18,Citation20,Citation21 To improve the accuracy of epitope prediction, HLA-A*0201 restricted peptides from CT23 protein sequence (NCBI Reference Sequence: NP_115878.2) were analyzed with various algorithms applied by seven different predictive programs ().
Table 1. Websites for epitope prediction.
CT23 candidate peptides, HLA-A*0201 positive control peptide GILGFVFTL (P+)Citation22 and HLA-A24 restricted epitope peptide KTPFVSPLL (P−)Citation23 were all synthesized by Hangzhou Dangang Biotechnology Co.,LTD. The purity of the peptides was over 95%. The dry powdered synthetic peptides were dissolved with DMSO, and then diluted to a concentration of 1 mg/mL with sterile deionized water. The diluted peptide solution was stored at −80°C for later use.
HLA-A2 binding affinity and stability of peptide/HLA-A2 complex
Due to the absence of TAP molecules, T2 cells (HLA-A2+, TAP−) cannot transport intracellular antigen peptides to HLA-I molecules. Consequently, HLA-A2 molecules on T2 cell surface are extremely unstable without antigen peptide binding and quickly degraded after expression. To take advantage of this feature of T2 cells, we loaded exogenous antigenic peptides on the surface of T2 cells to increase the stability of HLA-A2. As the binding force of antigen peptide and HLA-A2 is positively correlated with expression of HLA-A2 molecules on T2 cell surface, HLA-A2 expression on T2 surface reflects the binding affinity of foreign antigen peptides to HLA-A2. In this experiment, T2 cell binding experiments were performed on CT23 candidate peptides predicted above, and the experimental steps were carried out with reference to literatures.Citation24,Citation25 T2 cells (1 × 106/mL) were incubated in the presence of 50 µg/mL peptide in RPMI-1640 medium supplemented with 3 µg/mL human β2-microglobulin (Sigma, USA) at 37°C in 5% CO2 for 18 h, and the following tests were performed, respectively. Meanwhile, a blank control group of T2 cells treated without peptide was set up.
The above cells were washed with PBS for three times and incubated with FITC-labeled HLA-A2 monoclonal antibody (eBioscience, USA) for 30 min. Mean fluorescence intensity (MFI) of the cells was detected by flow cytometry. After that, the fluorescence index (FI) was used as an index to measure the binding force of the peptide and HLA-A2 molecule: FI = (MFI of peptide-loaded experimental group – MFI of blank control group)/MFI of blank control group. Moreover, FI > 1.0 indicates high binding affinity, 1.0>FI > 0.5 indicates medium binding affinity, and FI < 0.5 indicates low binding affinity.
The above cells were collected and washed twice to remove free peptides, then incubated with 10 μg/mL Brefeldin A (Sigma, USA) for 1 h, washed, and further cultured in RPMI-1640 medium at 37°C and 5% CO2. Cells were collected for each period (0, 2, 4, 6 and 8 h). Subsequently, cells were incubated with FITC-labeled HLA-A2 monoclonal antibody for 30 min, and MFI of each group was detected by flow cytometry. The results were represented by dissociation complex 50 (DC50), defined as the time required for the loss of 50% of the stabilized peptide/HLA-A2 complexes at time 0 h. Peptides with a high DC50 are considered to be more stable when they bind to HLA-A2.
Identification of HLA phenotype of tumor cells
Tumor cells in the logarithmic growth phase were collected. Later, the whole genome DNA of tumor cells was extracted by DNA extraction kit (TIANGEN, China), and sent to Shenzhen Blood Bank for HLA-A genotype identification.
Immunoblot
Total protein of tumor cells was extracted and then mixed with SDS-PAGE protein loading buffer, heated at 100°C for 5 mins until protein denaturation. After SDS-PAGE electrophoresis, the protein was transferred to the PVDF membrane, and then it was blocked with 5% skim milk powder solution for 2 h. After that, PVDF membrane was added with GAPDH antibody (1:1000) and CT23 antibody (1:500; Muliti sciences, China), incubated overnight at 4°C. After rinsing three times with PBST, PVDF membrane was incubated in horseradish peroxidase (HRP) labeled secondary antibodies (1:5000) at room temperature for 2 h. Finally, ECL chemiluminescence was used to visualize the PVDF membrane.
Generation of DC pulsed with peptides
DC induction in vitro was performed in accordance with the procedures previously described.Citation17 Briefly, peripheral blood mononuclear cells (PBMCs) from HLA-A2 healthy donor were prepared by Ficoll density gradient centrifugation. Then CD14+ cells (monocytes) were separated from PBMCs by Human CD14+ Cell Isolation Kit (Stemcell, Canada). The monocytes were cultured in RPMI-1640 medium with rhGM-CSF (100 ng/mL) and rhIL-4 (50 ng/mL), which were purchased from Peprotech, USA. Medium and cytokines were replaced half every other day. rhTNF-α (50 ng/mL) (Peprotech, USA) was added on the 5th day of culture, and the culture was continued until the 8th day, and mature DCs were harvested. Later, mature DCs were seeded in 24-well plates (1 × 105 cells/well) with peptide (50 μg/mL) or CT23 recombinant protein (100 μg/mL), respectively, grouped as follows: P(1–7)-DC group: P1-P7 candidate peptide was co-incubated with DCs; CT23-DC group: CT23 recombinant protein was co-incubated with DCs; P+-DC group: HLA-A*0201-restricted control peptide was co-incubated with DCs; P−-DC group: HLA-A24-restricted control peptide was co-incubated with DCs; NP-DC group: No peptide was co-incubated with DCs. After loading with peptides, DCs were cultured continuously for 4 h and collected for subsequent experiments.
Flow cytometry
DCs were collected on the 5th and 8th day after culture, washed twice with PBS, and added monoclonal antibody (anti-HLA-DR, anti-CD1a, anti-CD80, anti-CD86 or anti-CD83) or isotype-matched control antibody (eBioscience, USA), respectively. Subsequently, cells were incubated at 4°C for 30 min, and washed three times with PBS. Finally, flow cytometry was used to detect the expression of HLA-DR, CD80, CD83, CD86 and CD1a molecules on DC surface, and the specific steps were referred to the literature.Citation17
Induction of peptide-specific CTLs in vitro
CD8+T cells were separated from PBMCs of HLA-A2 healthy donor by EasySep™ Human CD8+T Cell Isolation Kit (Stemcell, Canada).Citation24 CD8+T cells were co-cultured with peptide-loaded DCs (CD8+T cells to DC ratio was 10:1). rhIL-2 (20 ng/mL) was added on the 2nd day, and the culture medium was changed in half every 2–3 days. On days 7 and 12 of culture, the same proportion of peptide-loaded DCs was added for enhanced stimulation.Citation26,Citation27 Cultured to day 15, cells were collected as effector T cells (i.e., CTLs).
T cells proliferation assay
Cell Counting Kit-8 (CCK-8,Shanghai Yeasen Biotechnology Co., Ltd.) assay was utilized to assess T Cells proliferation, according to manufacturer’s instructions. DCs and CD8+T cells were co-cultured at day 0 and day 14, cell suspensions of 20 μL each were placed in triplicate wells of a 96-well plate. Meanwhile, a blank control group containing 20 μL medium was set. 80 μL of complete media of PMI-1640 and 10 μL of CCK-8 solution were added to each well, and then cultured at 37°C and 5% CO2 for 3 h, and the optical density value (OD450) of each well was detected. The stimulation index (SI) of CD8+T cells in each group was calculated according to the following formula: SI= (OD450 value of day 14 experimental well – OD450 value of blank control well)/(OD450value of day 0 experimental well – OD450 value of blank control well).
Enzyme-linked immunosorbent assay (ELISA)
DCs and CD8+T cells co-cultured supernatant at day 3 and day 12 was taken, and content of IFN-γ in culture supernatant was detected by human IFN-γ ELISA kits (Cloud-Clone Corporation, Wuhan, China). Except for blank control, each well was mixed with 50 μL supernatant or different concentration standards and 100 μL HRP labeled antibody. After incubation at 37°C for 60 minutes, the plate was washed five times. 50 μL of substrate A and substrate B were added, respectively, and it was incubated at 37°C for 15 minutes. Next, 50 μL stop solution was added and the absorbance value (OD450) was measured within 15 minutes. Standard curve was drawn according to standard substance concentration and OD value, and IFN-γ concentration in supernatant was calculated.Citation24
Enzyme-linked immunospot assay (ELISPOT)
Firstly, CTLs induced by peptide-loaded DCs were collected, and IFN-γ secreting cell number was detected by Human IFN-γ ELISPOT kit (Dakewe, Beijing, China). CTLs of the above groups was added into the 96-well ELISPOT plate coated with anti-human IFN-γ antibody (1 × 105cells/well), and positive control (plus PHA) and background control wells were set according to the kit instructions. After incubating CTLs in a 37°C, 5% CO2 incubator for 20 hours, biotinylated antibody, streptavidin antistin protease conjugate and enzyme substrate nitro tetrazole were added to the wells for IFN-γ secreting cell detection according to the kit instructions. ELISPOT analyzer was used to scan the spots present in each well, and ImmunoSpot Professional software was used to count them.Citation24
Cytotoxicity assay
CytoTox 96® Non-radioactive Cytotoxicity Assay (Promega, USA) was used to detect the killing of tumor cells or T2 cells by lactate dehydrogenase (LDH) release. Tumor cells U87 (HLA-A *0201+,CT23+), OVCAR-3 (HLA-A *0201+, CT23+), SKOV3 (HLA-A *0201−, CT23+), T2 cells loaded with specific peptides and T2 cells without loading peptides were used as target cells. CTLs (effector cells) and target cells (50:1) were in mixed culture act as experimental wells. Meanwhile, effector cell spontaneous LDH release wells,target cell spontaneous LDH release wells and target cell maximum LDH release wells were established. After cultivation for 4 h, 50 μL supernatant from each well was taken and transferred to a new 96-well plate adding the substrate buffer (50 μL/well) and mixing it well. The mixture was incubated at room temperature without light for 30 min. 50 μL termination solution was added to terminate the reaction. The value of OD490 was measured by microplate reader and the specific killing rate of CTLs from each group was calculated by the following formula: killing rate (%) = (OD value of experimental wells – OD value of effector cell spontaneous release wells – OD value of target cell spontaneous release wells)/(OD value of target cell maximum release wells – OD value of target cell spontaneous release wells) ×100%.Citation20,Citation28,Citation29
Statistic analysis
The test results were set up in Excel database. Measurement data were represented by mean±standard deviation. Comparison between two sample means was performed by T test, and comparison between multiple sample means was performed by ANOVA.
Results
Prediction of and selection of HLA-A*0201 restricted epitopes of CT23
HLA-A*0201 restricted epitopes of CT23 were predicted using seven T cell epitope prediction tools. The higher scores indicate stronger binding to HLA-A*0201 in BIMAS, SYFPEITHI, NetCTL and NetChop prediction programs. And rank% was used in IEDB, NetMHC and NetMHCpan prediction programs to depicted binding status, in which the rank% of predicted peptide less than 0.5 was defined as strong binder and between 0.5 and 2 was defined as weak binder. Based on these, seven candidate peptide epitopes (named as P1-P7) were selected (). Then, Blast alignment confirmed that these peptide segments matched the amino acid sequence of CT23 (data not shown).
Table 2. In silico analysis of CT23 epitopes restricted to HLA-A*0201.
Binding affinity of CT23 candidate peptides to HLA-A2 and peptide/HLA-A2 complex stability
Since binding force between antigen peptides and HLA-A2 molecules on T2 cell surface is positively correlated with HLA-A2 expression on T2 cell surface, measurement of HLA-A2 expression on T2 cells surface will reflect the binding capability of peptides to HLA-A2 molecule after co-incubation of CT23 peptides with T2 cells. Results of the binding affinity and stability of CT23 peptides with T2 cell surface HLA-A2 molecules were shown in and . Peptide P1, P2, P4, P5, P6, and P7 exhibited higher binding affinity to HLA-A2 molecules, among which peptide P7 showed the highest binding affinity. And the binding affinity of peptide P3 to HLA-A2 molecules was moderate. The stability of peptides binding to HLA-A2 molecules was reflected by DC50. Except for peptide P2 and P6 DC50 less than 6 hours, remaining peptides had the DC50 greater than 6 hours, suggesting better stability in binding to HLA-A2 molecules.
Figure 1. Affinity and stability of CT23 candidate peptides binding to HLA-A2 analyzed by flow cytometry.
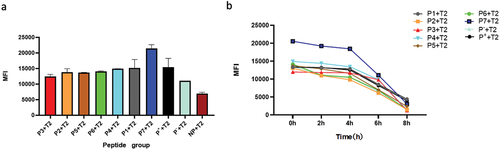
Table 3. The HLA-A2 binding affinity and stability of CT23 candidate peptides and control peptides.
Confirmation of HLA-A phenotype and CT23 expression in tumor cells
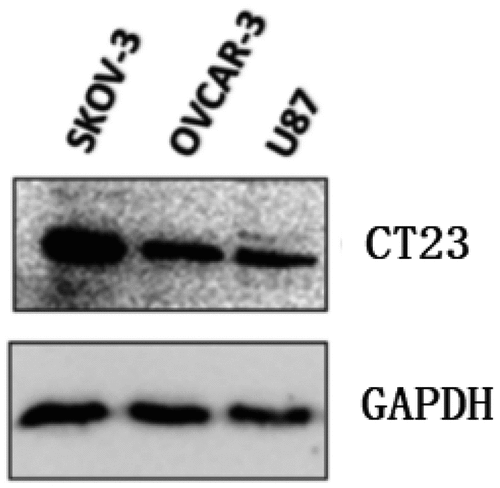
The HLA-A genotype analysis of tumor cell lines revealed that U87 had HLA-A alleles: A 02:01:01:01 and A 02:01:01:01; OVCAR-3 had HLA-A alleles: A 02:01:01:01 and A 29:02:01:01, confirming that both U87 and OVCAR-3 were HLA-A*0201 positive cells. Whereas SKOV3 had HLA-A alleles: A*03:01:01:01 and A 68:01:02:01, confirming that SKOV3 was HLA-A *0201 negative (data not shown). Finally, CT23 protein was detected in U87, SKOV3, and OVCAR-3 cells by immunoblot ().
Figure 2. Expression of CT23 protein in tumor cells detected by immunoblot.
DC identification
Morphological features of DC
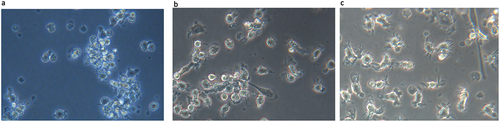
CD14+ cells were cultured in RPMI-1640 complete medium containing rhGM-CSF and rhIL-4 and differentiated into DCs. The maturation of DCs was induced by adding rhTNF-a on the 5th day of culture. The DCs at different culture periods were shown in : on the 3rd day of culture, the DCs grew from adherent status to semi-suspension status aggregating as clusters. The cell morphology was approximately round and no obvious protrusions on the surface (). On the 5th day of culture, cells were more dispersed with increasing volume, and some of them showed protrusions (). When cultured to the 8th day, the cells became large and translucent with various cell morphologies, for example, leaf like or stellate. The cell surface emerged dendritic protrusions with irregular shapes, different lengths and thicknesses, which was consistent with the morphological characteristics of mature DC ().
Figure 3. Morphological features of DCs in different stages.
Molecular phenotype of DC
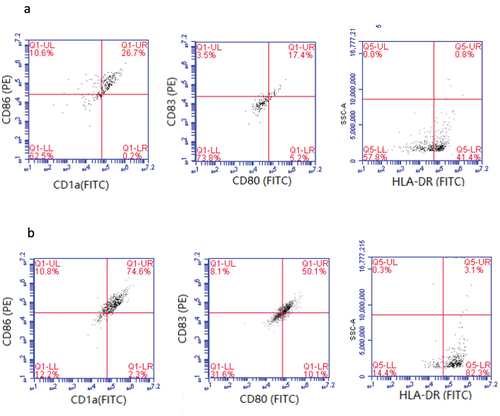
The surface molecules (HLA-DR, CD80, CD83, CD86 and CD1a) of DCs cultured for 5 and 8 days were detected by flow cytometry. The results are shown in : DCs cultured for 5 days presented low expression rates of CD1a (26.9%), CD86 (37.3%), CD80 (22.6%), CD83 (20.9%), and HLA-DR (42.2%) (), suggesting DCs were in immature stage. When cultured to day 8, cells showed 76.9% of CD1a, 85.4% of CD86, 60.2% of CD80, 58.2% of CD83, and 85.4% of HLA-DR (), which were significantly increased compared with those on day 5, indicating DCs had matured at that time.
Figure 4. The surface molecules of DCs detected by flow cytometry.
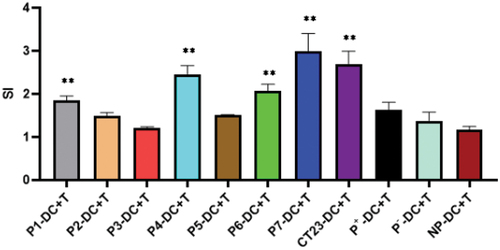
CD8+ T cell proliferation stimulated by peptide-loaded DCs
After the co-culture of CD8+T cells and DCs was loaded with antigenic peptides, CD8+T cells grew in clusters, and some showed division (data not presented), indicating that CD8+T cells showed proliferation signs after DCs stimulation. As demonstrated in , CD8+T cells stimulated by DCs loaded with peptide P1, P4, P6 and P7 or CT23 recombinant protein showed significantly higher SI, and SI in P7-DC+T group was the highest, which was statistically significant compared with the control group (NP-DC-T group) (p < .01). This result suggested that CD8+T can be activated by DC loaded with CT23 peptide or CT23 recombinant protein.
Figure 5. Proliferation assay of D8+T cell stimulated by peptide-loaded DCs.
Induced CTL with IFN-r production ability
Considering ability of CTLs to secrete IFN-γ, CD8+T cell activation was evaluated by testing IFN-γ content in culture medium and counting the number of IFN-γ-positive cells in CD8+T cells. Firstly, the culture medium from the 12th day of co-cultivation of DCs and CD8+T cells was collected, and IFN-γ content in each group was measured via ELISA analysis (). The results indicated that, except for the P3-CTL group, all other group showed increased IFN-γ secretion in various degrees. Significantly, P7-CTL group showed the highest IFN-γ secretion, followed by P1-CTL, P4-CTL and P6-CTL groups. These differences were statistical significance compared with NP-CTL (the control group).
Figure 6. IFN-γ production ability of CT23 peptide-induced CTLs.
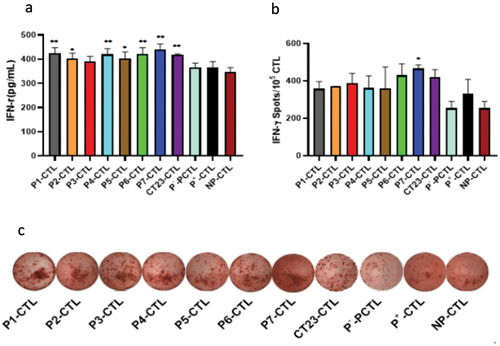
After that, the count of IFN-γ-positive cells was determined using ELISPOT, and the results are presented in . It was found that although the number of IFN-γ-positive cells increased in all groups compared to NP-CTL (the control group), only the P7-CTL group showed a significant statistical difference (p < .05).Taken together, above results demonstrated that CTLs induced by peptide P1-, P4-, P6- and P7 exhibited a stronger capacity to produce IFN-γ compared to other peptides of CT23, implying peptide P1-, P4-, P6- and P7 had superior ability in activating in CD8+T cells.
Induced CTL with CT23 specific cytotoxicity in vitro
Based on the experimental results presented above, we further conducted the test to determine whether P1, P4, P6, and P7-induced CTLs could kill tumor cells. As depicted in , the killing rate of P7 peptide-induced CTLs on HLA-A *0201+ tumor cells U87 and OVCAR-3 was found to be (31.42 ± 0.85)% and (31.81 ± 9.82)% respectively, which were significantly higher (p < .01) compared to control group (NP-CTL group). In contrast, although the killing rate of HLA-A *0201+ tumor cells by P1, P4, or P6-induced CTLs was increased, it was not statistically significant compared to the control group (NP-CTL group). Furthermore, CT23 peptide-induced CTLs did not demonstrate any killing effect on HLA-A*0201 negative SKOV3 tumor cells.
Figure 7. Cytotoxic activity of CT23 peptide-induced CTLs to target cells detected by LDH release.
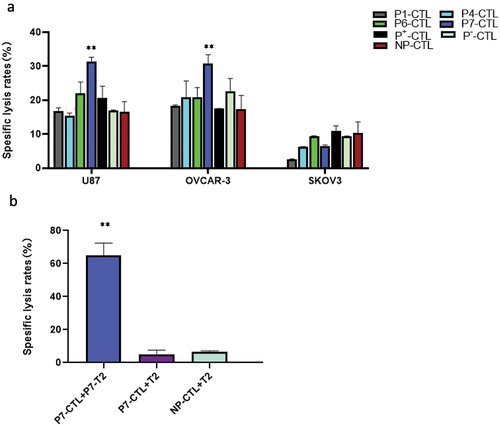
Given that P7-induced CTLs had higher killing effect on tumor cells, we next confirmed whether these CTLs were P7 specific. Applying peptide P7-loaded T2 cells as the target cells, we co-cultured these target cells with P7-induced CTLs and then determined the killing effect. The result revealed that P7-induced CTLs exhibited a significant killing effect on T2 cells loaded with peptide P7, with a remarkably high killing rate (64.95 ± 6.09)%. However, they did not exhibit any significant killing effect on T2 cells without loaded peptide P7. This result further confirmed that the induced CTLs were indeed P7 specific.
Discussion
Cancer remains a major health threat worldwide. In recent years, tumor immunotherapy has made significant progress despite encountering numerous challenges. Tumor antigens produced by tumor cells are processed and presented by antigen presenting cells, which result in activation of T cells and initiation of adaptive immune responses. Among these T cells, CD8+T cells can specifically kill tumor cells after activation into CTLs that are the main effector cells of body’s anti-tumor immunity. CTLs mediate the necrosis or apoptosis of tumor cells through the release of perforin, granzyme and tumor necrosis factor, and high expression of FasL, and so on. CTLs can effectively and specifically eliminate tumor cells and are usually considered as the first choice of immune cells for targeting tumors.Citation30 However, tumor cells usually have low expression of major histocompatibility complex (MHC) molecules known as human leukocyte antigens (HLA), and co-stimulatory molecules, and high expression of inhibitory molecules such as programmed death-ligand 1(PD-L1). The dysregulation of these molecules results in inhibition of the activation and the effect of CD8+ T cells in vivo, so that tumor immune escape occurs.Citation31–33 Numerous studies have claimed that after being cultured and activated in vitro, tumor-specific CTLs then infused back into tumor patients and can better kill tumor cells. Currently, CTLs are still one of the main research directions of adoptive cell immunotherapy.Citation4,Citation5,Citation30
Previous studies have reported that CT23 is not only highly expressed in various tumors, but also presents detectable specific antibodies in the serum of tumor patients, while the serum of healthy individual is negative.Citation9,Citation13–15,Citation17 These results suggest that CT23 is immunogenic like many other CTAs. Therefore, in recent years, many scholars have explored the possibility of CT23 as a target for tumor immunotherapy. In one of the studies, Luo et al.Citation17 reported that human-derived DCs loaded with recombinant CT23 protein were used to activate specific CTLs in vitro, and these specific CTLs could kill human hepatocellular carcinoma cells expressing CT23. Safavi et al.Citation34,Citation35 selected the most immunodominant epitopes of CT23 and synaptonemal complex protein 1 (SYCP1) to construct a novel polyepitope peptide vaccine that was applied in the murine model of melanoma and triple-negative breast cancer, respectively. They demonstrated that this polyepitope peptide vaccine has high efficacy for the immune system activation and anti-tumor prophylactic effects. In another study, CT23-transducing DCs were selected to stimulate T cells. The mice experiments both in vivo and in vitro showed that these T cells could produce specific anti-tumor immune effects, and when combined with epigenetic drugs, the anti-tumor effects of specific T cells could be enhanced due to up-regulation of CT23 in tumor cells.Citation36 In addition, Okumura et al.Citation23 identified an HLA-A24-restricted CT23 epitope and confirmed that the CTLs activated by this epitope peptide could specifically kill tumor cells (CT23+, HLA-A24+). These findings indicated the potential application prospect of CT23 in tumor immunotherapy.
DCs are the most functional antigen-presenting cells in human body and the only cells that can present antigens to activate naive T cell, thereby initiating the primary immune response. DCs can process and present tumor antigens to CD8+T cells or CD4+T cells through MHC-I or MHC-II presentation pathway, respectively, thus activating tumor-specific cellular immune response. At present, substantial evidence has confirmed that induced culture of DCs with tumor antigen in vitro can enhance antigen-presenting ability of DCs, activate CD8+T cells and reinforce the tumor-specific killing function of CTLs. Currently, application of DCs-CTLs not only has been proved safe and effective in malignant tumor treatment, but also improve patient’s autoimmune function, generate immune memory protection, and obtain a long-term anti-tumor effect.Citation37–41
In DC-CTL immunotherapy, how the antigen delivered to DCs is an important factor affecting immune efficacy. Various methods can be employed to load antigens onto DCs, including synthetic peptide/protein-based vaccines, DNA vaccines, mRNA vaccines, and others. Among these approaches, DNA vaccines have the potential to induce both cellular and humoral immune responses. However, it is important to consider certain drawbacks associated with DNA integration into the host genome, such as an increased risk of genomic alteration and the possibility of autoimmune responses against DNA, which may lead to autoimmune diseases.Citation6 On the other hand, mRNA vaccines offer several advantages. They are known for their rapid development, relatively low manufacturing costs, and safe administration. However, there are still some aspects regarding mRNA vaccine translation efficiency, mRNA stability, and the precise mechanism of delivery to the innate immune system for cancer therapy that require further elucidation.Citation3 Another approach for loading antigen onto DCs is through synthetic peptides. This method offers several advantages, such as ease of preparation, purification, transportation, and preservation, leading to well-controlled contamination. Additionally, synthetic peptides are more stable than proteins. Therefore, tumor antigen peptide-loaded DCs vaccines hold promising prospects for tumor immunotherapy.Citation42–44 In this study, we initially utilized the online databases to predict and screen seven CT23-epitopes with HLA-A*0201 restriction. Subsequently, we determined that these candidate epitope peptides were CT23-specific through Blast search comparison. Then, we further confirmed that these candidate peptides had good binding power and stability to HLA-A2 molecules through T2 cell binding assay. While the T2 cell binding assay has been widely used in similar studies,Citation24,Citation25,Citation45 it should be noted that it does not directly confirm peptide-HLA stability. Therefore, we conduct several complementary assays to address this limitation. Furthermore, as peptide-HLA stability alone may not guarantee immunogenicity, we further validated the ability of these peptides to activate CD8+T cells. By isolating CD14+ monocytes from peripheral blood, inducing them to become DCs loaded with CT23 candidate peptides, and using the DCs to stimulate autologous CD8+T cells, we demonstrated that all CT23 candidate peptides had the ability to activate and stimulate the proliferation of CD8+T cells. Among these peptides, peptide P7 loaded DCs exhibited the most significant activation of CD8+T cells, as evidenced by the highest levels of IFN-γ secretion and the greatest number of positive cells. It is worth noting that the P7-induced CTLs did not exhibit any killing effect on HLA-A*0201 negative tumor cells, indicating that the killing effect is restricted to HLA-A*0201. Moreover, P7-induce-CTLs could kill T2 cells loaded with peptide P7, but had no killing effect on T2 cells without loading the peptide P7, further indicating that P7 induced-CTLs were P7-specific. Based on the experimental results of these seven CT23 candidate peptides, peptide P7 (ALLVLCYSI) shows superior immunogenicity and holds prospects in application of tumor immunotherapy.
CTLs are able to kill tumor cells by recognizing tumor antigenic peptides presented by MHC-I molecules. However, tumor cells circumvent surveillance and elimination of immune cells including CTLs through a series of strategies. A key mechanism of cancer immune evasion is down-regulation of MHC-I that is responsible for the presentation of cancer antigenic peptides to CTLs. Numerous studies report that loss of MHC-I occurs frequently in human tumors, which is associated with worse clinical outcome and is also one of crucial influence factors lead to resistance of tumor immunotherapy based on CD8+T cells.Citation46,Citation47 In this study, we confirmed that P7-induced-CTLs can specifically kill CT23-positive tumor cells, but the killing efficiency seems to be lower, at approximately 31%. This result was similar to the killing efficiency of CT23 HLA-A24-restricted CTLs (CT23+, HLA-A24+) reported by Okumura et al.Citation23 However, when T2 cells loaded with the peptide P7 were used as target cells, the killing efficiency of P7-induced CTLs significantly increased (64.95 ± 6.09%). We believe that this difference in the killing effect of CTLs might not caused by experimental technique issue as all experimental conditions were the same and under strict control. The reason behind the lower killing efficiency in the CTLs targeting tumor cells may be due to the low expression of MHC-I molecules on the surface of the tumor cells. On the other hand, other factors contributing to the low killing efficiency of CTLs against tumor cells also can be considered. For example, the heterogeneity of tumor antigen expression, secretion of immune-suppressive molecules by tumor cells (such as TGF-β, IL-10, etc.), high expression of various anti-apoptotic molecules (such as Bcl-2), lack or weak expression of apoptosis-inducing molecules like Fas, etc. All these factors can contribute to the reduced killing ability of CTLs.
Despite the fact that many mechanisms for loss of MHC-I remain unclear, several methods for restoring MHC-I expression have been found and explored in anti-tumor treatment,Citation32,Citation48 e.g. the use of IFN-γ, Morrison et al.Citation49 showed that the increase of MHC-I expression on lung cancer cells treated with IFN-γ was able to partially sensitize tumor cells to T-cell-mediated recognition and lysis. Di Tomaso et al.Citation50 demonstrated that tumor stem cells from glioblastoma in the presence of IFN-γ also displayed increased MHC-I expression. Similarly, the report by Liao et al.Citation51 indicated that the expression of MHC-I can be up-regulated by IFN-γ treatment in cancer stem cells leading to improvements in T-cell recognition and lysis. Another method is the application of epigenetic drugs. In a previous study by our group, we explored epigenetic drugs (decitabine, trichostatin A and so on) to up-regulate MHC and MAGE-D4 that is a type of glioma associated antigen, which resulted in enhancing CTLs’ lysis to glioma cells.Citation24 Likewise, Luo et al.Citation52 demonstrated that hypomethylating agent, guadecitabine, upregulated MHC-I to potentiate CTLs responses in breast cancer. In the report of Ritter et al., they exhibited that histone deacetylases (HDACs) inhibitor vorinostat could restored HLA class-I surface expression in vitro and in a mouse xenotransplantation model on Merkel cell carcinoma.Citation53 Recently, there is a report about MHC class I and sumoylation that is a post-translational modification characterized by covalent and reversible binding of small ubiquitin-like modifier (SUMO) to a target protein. The researchers demonstrated that activated sumoylation permitted cancer cells to evade CD8+T cell–mediated immunosurveillance by suppressing the MHC-I antigen-processing and presentation machinery. And the use of the pharmacological inhibitor of SUMOylation, TAK-981, enhanced the presentation of antigens and the susceptibility of tumor cells to CD8+T cell – mediated killing.Citation54 In view of above promising research results, we propose that the combination of CT23 peptide induced CTLs and modulation of MHC I expression by diverse strategies to augment tumor immunogenicity could improve MHC I dependent immunotherapeutic efficacy, which could also be of therapeutic interest in future.
In this study, we have successfully demonstrated the specific killing ability of CTLs stimulated by DCs loaded with the CT23 peptide P7 (ALLVLCYSI) against tumor cells expressing CT23. Furthermore, we observed that this killing activity was restricted by HLA-A*0201. However, it is important to acknowledge certain limitations in our study, such as the lack of direct evidence regarding peptide-HLA stability and the absence of spontaneous peptide presentation by patients. Therefore, further validating the potential of the peptide P7 as a promising candidate for tumor-specific CTL therapy is warranted.
Author contributions
Rong Fan and Xiao-xun Xie were involved in the conception and design. xiao-qiong Zou,Feng Li,Ying-Ying Ge,Qing-Mei Zhang,Bin Luo,Wei Huang; Jian-Xia Zou were involved in the analysis and interpretation of the data. Xia Zeng,Wei-Xia Nong and Xiao-xun Xie were involved in the drafting of the paper, revising it critically for intellectual content, and the final approval of the version to be published. All authors agree to be accountable for all aspects of the work.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Salmaninejad A, Zamani MR, Pourvahedi M, Golchehre Z, Hosseini BA, Rezaei N. Cancer/testis antigens: expression, regulation, tumor invasion, and use in immunotherapy of cancers. Immunol Invest. 2016;45(7):619–12. doi:10.1080/08820139.2016.1197241.
- Gibbs ZA, Whitehurst AW. Emerging contributions of cancer/testis antigens to neoplastic behaviors. Trends Cancer. 2018;4(10):701–712. doi:10.1016/j.trecan.2018.08.005.
- Fan C, Qu H, Wang X, Sobhani N, Wang L, Liu S, Xiong W, Zeng Z, Li Y. Cancer/testis antigens: from serology to mRNA cancer vaccine. Semin Cancer Biol. 2021;76:218–31. doi:10.1016/j.semcancer.2021.04.016.
- Meng X, Sun X, Liu Z, He Y. A novel era of cancer/testis antigen in cancer immunotherapy. Int Immunopharmacol. 2021;98:107889. doi:10.1016/j.intimp.2021.107889.
- Nin DS, Deng LW. Biology of cancer-testis antigens and their therapeutic implications in cancer. Cells. 2023;12(6):926. doi:10.3390/cells12060926.
- Ren S, Zhang Z, Li M, Wang D, Guo R, Fang X, Chen F. Cancer testis antigen subfamilies: attractive targets for therapeutic vaccine (review). Int J Oncol. 2023;62(6):71. doi:10.3892/ijo.2023.5519.
- Jäger E, Gnjatic S, Nagata Y, Stockert E, Jäger D, Karbach J, Neumann A, Rieckenberg J, Chen YT, Ritter G, et al. Induction of primary NY-ESO-1 immunity: CD8+ T lymphocyte and antibody responses in peptide- vaccinated patients with NY-ESO-1+ cancers. Proc Natl Acad Sci USA. 2000 Oct 24;97(22):12198–203. doi:10.1073/pnas.220413497.
- Marchand M, van Baren N, Weynants P, Brichard V, Dréno B, Tessier MH, Rankin E, Parmiani G, Arienti F, Humblet Y, et al. Tumor regressions observed in patients with metastatic melanoma treated with an antigenic peptide encoded by gene MAGE-3 and presented by HLA-A1. Int J Cancer. 1999 Jan 18;80(2):219–30. doi:10.1002/(SICI)1097-0215(19990118)80:2<219:AID-IJC10>3.0.CO;2-S.
- Ono T, Kurashige T, Harada N, Noguchi Y, Saika T, Niikawa N, Aoe M, Nakamura S, Higashi T, Hiraki A, et al. Identification of proacrosin binding protein sp32 precursor as a human cancer/testis antigen. Proc Natl Acad Sci USA. 2001;98(6):3282–7. doi:10.1073/pnas.041625098.
- Whitehurst AW, Xie Y, Purinton SC, Cappell KM, Swanik JT, Larson B, Girard L, Schorge JO, White MA. Tumor antigen acrosin binding protein normalizes mitotic spindle function to promote cancer cell proliferation. Cancer Res. 2010;70(19):7652–61. doi:10.1158/0008-5472.CAN-10-0840.
- Fu J, Luo B, Guo WW, Zhang QM, Shi L, Hu QP, Chen F, Xiao SW, Xie XX. Down-regulation of cancer/testis antigen OY-TES-1 attenuates malignant behaviors of hepatocellular carcinoma cells in vitro. Int J Clin Exp Pathol. 2015;8:7786–7797.
- Ning W, Huang M, Wu S, Wang H, Yao J, Ge Y, Tang Y, Sun K, Xie X, Hu Q. CT23 knockdown attenuating malignant behaviors of hepatocellular carcinoma cell is associated with upregulation of metallothionein 1. Cell Biol Int. 2021;45(6):1231–1245. doi:10.1002/cbin.11567.
- Lin L, Nong W, Luo B, Ge Y, Zeng X, Li F, Fan R, Zhang Q, Xie X. Cancer-testis antigen ACRBP expression and serum immunoreactivity in ovarian cancer: its association with prognosis. Immun Inflamm Dis. 2021;9(4):1759–1770. doi:10.1002/iid3.534.
- Li X, Yan J, Fan R, Luo B, Zhang Q, Lin Y, Zhou S, Luo G, Xie X, Xiao S. Serum immunoreactivity of cancer/testis antigen OY-TES-1 and its tissues expression in glioma. Oncol Lett. 2017;13(5):3080–6. doi:10.3892/ol.2017.5799.
- Luo B, Yun X, Fan R, Lin YD, He SJ, Zhang QM, Mo FR, Chen F, Xiao SW, Xie XX. Cancer testis antigen OY-TES-1 expression and serum immunogenicity in colorectal cancer: its relationship to clinicopathological parameters. Int J Clin Exp Pathol. 2013;6:2835–2845.
- Fan R, Huang W, Luo B, Zhang QM, Xiao SW, Xie XX. Cancer testis antigen OY-TES-1: analysis of protein expression in ovarian cancer with tissue microarrays. Eur J Gynaecol Oncol. 2015;36:298–303.
- Luo B, Yun X, Li J, Fan R, Guo WW, Liu C, Lin YD, Ge YY, Zeng X, Bi SQ, et al. Cancer-testis antigen OY-TES-1 expression and immunogenicity in hepatocellular carcinoma. Curr Med Sci. 2020;40(4):719–728. doi:10.1007/s11596-020-2241-x.
- Feola S, Chiaro J, Martins B, Cerullo V. Uncovering the tumor antigen landscape: what to know about the discovery process. Cancers Basel. 2020 Jun 23;12(6):1660. doi:10.3390/cancers12061660.
- Liang B, Zhu L, Liang Z, Weng X, Lu X, Zhang C, Li H, Wu X. A simplified PCR-SSP method for HLA-A2 subtype in a population of Wuhan, China. Cell Mol Immunol. 2006;3:453–458.
- Tang B, Zhou W, Du J, He Y, Li Y. Identification of human leukemia antigen A*0201-restricted epitopes derived from epidermal growth factor pathway substrate number 8. Mol Med Rep. 2015;12(2):1741–1752. doi:10.3892/mmr.2015.3673.
- Bonsack M, Hoppe S, Winter J, Tichy D, Zeller C, Küpper MD, Schitter EC, Blatnik R, Riemer AB. Performance evaluation of MHC class-I binding prediction tools based on an experimentally validated MHC-peptide binding dataset.Cancer Immunol Res. 2019;7(5):719–736. doi:10.1158/2326-6066.CIR-18-0584.
- Rosendahl Huber SK, Luimstra JJ, van Beek J, Hoppes R, Jacobi RH, Hendriks M, Kapteijn K, Ouwerkerk C, Rodenko B, Ovaa H, et al. Chemical Modification of Influenza CD8+T-Cell Epitopes Enhances Their Immunogenicity Regardless of Immunodominance. PLoS ONE. 2016 Jun 22;11(6):e0156462. doi:10.1371/journal.pone.0156462.
- Okumura H, Noguchi Y, Uenaka A, Aji T, Ono T, Nakagawa K, Aoe M, Shimizu N, Nakayama E. Identification of an HLA-A24-restricted CT23 epitope recognized by cytotoxic T cell. Microbiol Immunol. 2005;49(11):1009–1016. doi:10.1111/j.1348-0421.2005.tb03688.x.
- Bi SQ, Zhang QM, Zeng X, Liu C, Nong WX, Xie H, Li F, Lin LN, Luo B, Ge YY, et al. Combined treatment with epigenetic agents enhances anti-tumor activity of MAGE-D4 peptide-specific T cells by upregulating the MAGE-D4 expression in glioma. Front Oncol. 2022 Aug 3;12:873639. doi:10.3389/fonc.2022.873639.
- Liu W, Zhai M, Wu Z, Qi Y, Wu Y, Dai C, Sun M, Li L, Gao Y. Identification of a novel HLA-A2-restricted cytotoxic T lymphocyte epitope from cancer-testis antigen PLAC1 in breast cancer. Amino Acids. 2012;42(6):2257–2265. doi:10.1007/s00726-011-0966-3.
- Butterfield LH, Meng WS, Koh A, Vollmer CM, Ribas A, Dissette VB, Faull K, Glaspy JA, McBride WH, Economou JS. T cell responses to HLA-A*0201-restricted peptides derived from human alpha fetoprotein. J Immunol. 2001;166(8):5300–5308. doi:10.4049/jimmunol.166.8.5300.
- Wu Y, Wan T, Zhou X, Wang B, Yang F, Li N, Chen G, Dai S, Liu S, Zhang M, et al. Hsp70-like protein 1 fusion protein enhances induction of carcinoembryonic antigen-specific CD8+ CTL response by dendritic cell vaccine. Cancer Res. 2005;65(11):4947–4954. doi:10.1158/0008-5472.CAN-04-3912.
- Chen Q, Jia G, Zhao X, Bao Y, Zhang Y, Ozkan C, Minev B, Ma W. Novel survivin peptides screened with computer algorithm induce cytotoxic T lymphocytes with higher cytotoxic efficiency to cancer cells. Front Mol Biosci. 2020 Sep 2;7:570003. doi:10.3389/fmolb.2020.570003.
- Li J, Bai J, Gu L, He A, Wang J, Wang J, Zhang P, Zhang W. Prediction and identification of HLA-A*0201-restricted epitopes from leukemia-associated protein MLAA-22 which elicit cytotoxic T lymphocytes. Med Oncol. 2014;31(12):293. doi:10.1007/s12032-014-0293-0.
- Farhood B, Najafi M, Mortezaee K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: a review. J Cell Physiol. 2019;234(6):8509–21. doi:10.1002/jcp.27782.
- Khandelwal N, Breinig M, Speck T, Michels T, Kreutzer C, Sorrentino A, Sharma AK, Umansky L, Conrad H, Poschke I, et al. A high-throughput RNAi screen for detection of immune-checkpoint molecules that mediate tumor resistance to cytotoxic T lymphocytes. EMBO Mol Med. 2015;7(4):450–463. doi:10.15252/emmm.201404414.
- Sari G, Rock KL. Tumor immune evasion through loss of MHC class-I antigen presentation. Curr Opin Immunol. 2023 Aug;83:102329. doi:10.1016/j.coi.2023.102329.
- Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer. 2021;21(5):298–312. doi:10.1038/s41568-021-00339-z.
- Safavi A, Kefayat A, Sotoodehnejadnematalahi F, Salehi M, Modarressi MH. Production, purification, and in vivo evaluation of a novel multiepitope peptide vaccine consisted of immunodominant epitopes of SYCP1 and ACRBP antigens as a prophylactic melanoma vaccine. Int Immunopharmacol. 2019;76:105872. doi:10.1016/j.intimp.2019.105872.
- Safavi A, Kefayat A, Mahdevar E, Ghahremani F, Nezafat N, Modarressi MH. Efficacy of co-immunization with the DNA and peptide vaccines containing SYCP1 and ACRBP epitopes in a murine triple-negative breast cancer model. Hum Vaccin Immunother. 2021;17(1):22–34. doi:10.1080/21645515.2020.1763693.
- Bi SQ, Zhang QM, Zeng X, Liu C, Nong WX, Xie H, Li F, Lin LN, Luo B, Ge YY, et al. Combined treatment with epigenetic agents enhances anti-tumor activity of T cells by upregulating the ACRBP expression in hepatocellular carcinoma. Am J Transl Res. 2021;13:7591–7609.
- Najafi S, Mortezaee K. Advances in dendritic cell vaccination therapy of cancer. Biomed Pharmacother. 2023;164:114954. doi:10.1016/j.biopha.2023.114954.
- Wang Y, Yang X, Yu Y, Xu Z, Sun Y, Liu H, Cheng J, Liu M, Sha B, Li L, et al. Immunotherapy of patient with hepatocellular carcinoma using cytotoxic T lymphocytes ex vivo activated with tumor antigen-pulsed dendritic cells. J Cancer. 2018 Jan 1;9(2):275–287. doi:10.7150/jca.22176.
- Wang H, Wang J, Wang Q, Yang Y, Guo J, Ren C, Mou Y, Jia C, Song X, Song X. Laryngeal extra-skeletal Ewing sarcoma treated with DC-CTL immunotherapy: a case report and review of the literature. Front Oncol. 2022 Nov 30;12:1003393. doi:10.3389/fonc.2022.1003393.
- Ren PT, Zhang Y. Comparative investigation of the effects of specific antigensensitized DCCIK and DCCTL cells against B16 melanoma tumor cells. Mol Med Rep. 2017;15(4):1533–1538. doi:10.3892/mmr.2017.6175.
- Wang Y, Xu Z, Zhou F, Sun Y, Chen J, Li L, Jin H, Qian Q. The combination of dendritic cells-cytotoxic T lymphocytes/cytokine-induced killer (DC-CTL/CIK) therapy exerts immune and clinical responses in patients with malignant tumors. Exp Hematol Oncol. 2015 Nov 10;4(1):32. doi:10.1186/s40164-015-0027-9.
- Zhong G, Zhao W, Li Y, Jin G, Zeng W, Yu C, Zhou J, Yu L. MAGEA1 and hTERT Peptide treatment improves the potency of the dendritic cell- cytotoxic T lymphocytes (DC-CTL) immunotherapy in DAC treated acute myeloid leukemia. J Cancer. 2022 Jan 24;13(4):1252–1260. doi:10.7150/jca.66501.
- Wei X, Chen F, Xin K, Wang Q, Yu L, Liu B, Liu Q. Cancer-testis antigen peptide vaccine for cancer immunotherapy: progress and prospects. Transl Oncol. 2019;12(5):733–8. doi:10.1016/j.tranon.2019.02.008.
- Liao F, Zhang J, Hu Y, Najafabadi AH, Moon JJ, Wicha MS, Kaspo B, Whitfield J, Chang AE, Li Q. Efficacy of an ALDH peptide-based dendritic cell vaccine targeting cancer stem cells. Cancer Immunol Immunother. 2022;71(8):1959–73. doi:10.1007/s00262-021-03129-6.
- Zeng JZ, Liu Y, Huang F, He ZH, Sun H, Lu YD, Lei JH, Luo RC. Glypican-3-specific cytotoxic T lymphocytes induced by human leucocyte antigen-A*0201-restricted peptide effectively kill hepatocellular carcinoma cells in vitro. Asian Pac J Trop Med. 2017 Nov;10(11):1084–9. doi:10.1016/j.apjtm.2017.10.013.
- Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol. 2021 Mar 9;12:636568. doi:10.3389/fimmu.2021.636568.
- Cornel AM, Mimpen IL, Nierkens S. MHC class I downregulation in cancer: underlying mechanisms and potential targets for cancer immunotherapy. Cancers Basel. 2020 Jul 2;12(7):1760. doi:10.3390/cancers12071760.
- Gocher AM, Workman CJ, Vignali DAA. Interferon-γ: teammate or opponent in the tumour microenvironment? Nat Rev Immunol. 2022;22(3):158–72. doi:10.1038/s41577-021-00566-3.
- Morrison BJ, Steel JC, Morris JC. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of cancer-initiating cells. BMC Cancer. 2018 Apr 26;18(1):469. doi:10.1186/s12885-018-4389-3.
- Di Tomaso T, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A, Mortini P, Ferrone S, Doglioni C, Marincola FM, et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res. 2010;16(3):800–813. CCR-09-2730. doi:10.1158/1078-0432.CCR-09-2730.
- Liao T, Kaufmann AM, Qian X, Sangvatanakul V, Chen C, Kube T, Zhang G, Albers AE. Susceptibility to cytotoxic T cell lysis of cancer stem cells derived from cervical and head and neck tumor cell lines. J Cancer Res Clin Oncol. 2013;139(1):159–70. doi:10.1007/s00432-012-1311-2.
- Luo N, Nixon MJ, Gonzalez-Ericsson PI, Sanchez V, Opalenik SR, Li H, Zahnow CA, Nickels ML, Liu F, Tantawy MN, et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat Commun. 2018 Jan 16;9(1):248. doi:10.1038/s41467-017-02630-w.
- Ritter C, Fan K, Paschen A, Reker Hardrup S, Ferrone S, Nghiem P, Ugurel S, Schrama D, Becker JC. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Sci Rep. 2017 May 23;7(1):2290. doi:10.1038/s41598-017-02608-0.
- Demel, UM, Böger M, Yousefian S, Grunert C, Zhang L, Hotz PW, Gottschlich A, Köse H, Isaakidis K, Vonficht D, et al. Activated SUMOylation restricts MHC class I antigen presentation to confer immune evasion in cancer. J Clin Invest. 2022;132(9):e152383. doi:10.1172/JCI152383.