
ABSTRACT
Auxin/Indole-3-Acetic Acid (Aux/IAA) genes are involved in auxin signaling pathway and play an important role in plant growth and development. However, many studies focus on Aux/IAA gene families and much less known in Bletilla striata. In this study, a total of 27 Aux/IAA genes (BsIAA1-27) were cloned from the transcriptome of Bletilla striata. Based on a phylogenetic analysis of the Aux/IAA protein sequences from B. striata, Arabidopsis thaliana and Dendrobium officinale, the Aux/IAA genes of B. striata (BsIAAs) were categorized into 2 subfamilies and 9 groups. While BsIAAs were more closer to those of D. officinale compared to A. thaliana. EST-SSR marker mining test showed that 4 markers could be stably amplified with obvious polymorphisms among 4 landraces. Our results suggested that BsIAAs were involved in the process of tuber development and provided insights into functional roles of Aux/IAA genes in B. striata and other plants.
Graphical Abstract
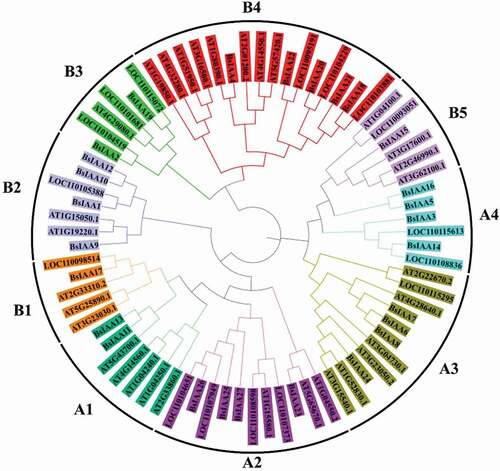
In this paper, we identified the 27 members of the Aux/IAA gene family which can be divided into 2 groups and 9 subfamilies. The similar branches of Aux/IAA proteins had the same or similar motifs, such as genes between BsIAA10 and BsIAA12, BsIAA11 and BsIAA13, BsIAA18 and BsIAA20, indicating that Aux/IAA proteins were conserved. Among them, BsIAAs were more closer to those of D. officinale compared to A. thaliana.
Introduction
Auxin is the first discovered plant hormone playing an important role in plant growth and development [Citation1, Citation2]. It consists of a group of molecules with an anthracene ring and is a commonly used signal chemical in plants. Auxin regulates cell division and elongation of plants, and organ development of cells and whole plants [Citation2]. Growing evidence indicates that auxin, either alone or together with other hormones, is involved in plant responses to environmental stimuli, including drought, cold, and salt [Citation3,Citation4]. Aux/IAA, SAUR and GH3 are the three major gene families of early auxin response, which are responsive to early auxin induction [Citation5]. As an important regulatory gene in the auxin signaling pathway, Aux/IAA gene has become a hot topic in recent years. Its protein sequence has four conserved domains. Domain I has transcriptional inhibition, II acts on the stability of its protein, III and IV are responsible for protein dimerization [Citation6]. This protein and the transcription factor ARF (auxin response factor) family can form heterodimers. Under the action of auxin, the protein of Aux/IAA is degraded, which triggers the expression of downstream genes of auxin signaling pathway [Citation7,Citation8]. Since the first Aux/IAA gene was cloned, 32 Aux/IAA genes have been identified from Arabidopsis genome-wide analysis, and a large number of Aux/IAA family members have also been identified in other plants, including eucalyptus [Citation9], cucumber [Citation10], maize [Citation11], soybean [Citation12] and so on. Genome-wide analysis showed that the members of the Aux/IAA gene family had different biological functions. In Hedychium coronarium, HcIAA2 and HcIAA4 play important roles in its floral scent formation [Citation13]. In Medicago truncatula, MtIAA6 and MtIAA7 exhibit root-specific expressions and MtIAA9 shows higher expression level in flower [Citation14]. In Dendrocalamus sinicus, DsIAA3, DsIAA4, DsIAA15 and DsIAA20 may be important for regulating shoot development [Citation15]. In conclusion, these studies indicate that the Aux/IAA gene family is involved in the regulation of plant growth and development and response to multiple signal transduction pathways. The analysis of Aux/IAA gene family is not only helpful to elucidate the molecular mechanism of auxin metabolism and signal transduction, but also can be used in plant genetic research.
Bletilla striata is a perennial herbaceous temperate plant of Orchidaceae with many significant values on medicine, ornamental and so on [Citation16]. And the secondary metabolites are the important medicinal components of it, so it is necessary to analyze the genes related to the synthesis of the secondary metabolites. However, there are few reports on its growth regulation and secondary metabolite synthesis. And there is no report on the systematic identification and analysis of Aux/IAA gene family in B. striata now. In consequence, this study intended to analyze the Aux/IAA gene family members of B. striata by bioinformatics methods based on the entire transcriptome data of developmental organs covering the entire growth phase in the early stage, and design specific molecular markers based on their sequences. It may provide a theoretical basis for the related utilization of Aux/IAA genes in B. striata, and provide clues for the study of functional characteristics of auxin-responsive genes.
1 Materials and methods
1.1 Materials
The B. striata seeds were purchased from a local farmer planting medicinal herbs in Zheng’an County (28°56′N, 107°43′E), Guizhou Province, China, on October 20th, 2015. The seeds were germinated and formed into seedlings by tissue culture, and the refined seedlings were transplanted to the test site. The protocorms, whole seedlings before seedling transplanting, whole seedlings after transplanting for 2 months, whole plants after transplanting for 1 year (including roots, stems, leaves, flowers, and seeds for successful pollination) were randomly collected for total RNA extraction. Then after the detection, the qualified RNA samples were pooled with the same amount for the subsequent transcriptome sequencing [Citation17].
1.2 Methods
1.2.1 Transcriptome assembly
Using the Illumina HiSeq sequencing 2000 platform to conduct high-throughput sequencing of B. striata.And the resulting data were assembled by de novo using Trinity software to finally obtain the transcriptome data set of B. striata’s single gene sequence.
1.2.2 Aux/IAA gene family identification
The sequences of Aux/IAA were obtained for identify the conserved domains. Aux/IAA genes of Arabidopsis (AtIAAs) were screened out by querying against the TAIR (The Arabidopsis Information Resource, http://www.arabidopsis.org/). Local tblastn search of B. striata proteomes by using Bioedit software (score value≥100 and evalue≤e−10) [Citation18]. Selecting the assembled data of our group as the search database, and using the 29 protein sequences of Arabidopsis as query [Citation19]. Using the online software Pfam and NCBI blast to screen for candidate sequences. All obtained protein sequences were examined for the presence of Aux/IAA (PF02309) domains by using the Hidden Markov Model of Pfam, SMART (http://smart.embl-heidelberg.de/) and InterPro (http://www.ebi.ac.uk/interpro/) tools.
1.2.3 Bioinformatics analysis of proteins of Aux/IAA genes
To predict the coding sequences, we first applied ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/) to predict the Open Reading Frame (ORF) of unigenes successfully matched by BLAST. Then, the basic physicochemical properties such as molecular weight, isoelectric point, and instability coefficient of each nucleotide sequence of BsIAAs were predicted by the online software ProtParam (http://web.expasy.org/protparam/). The subcellular localization of BsIAA proteins was analyzed using the online software Plant-mPLoc server (http://www.csbio.sjtu.edu.cn/bioinf/plant-multi/#). The secondary structure was analyzed using the online server SOPMA (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html).
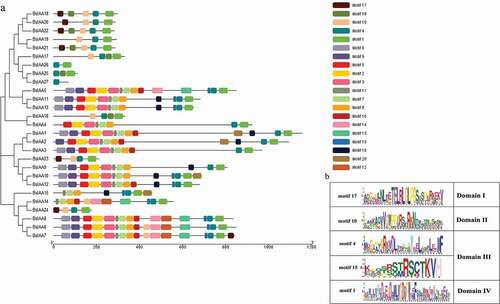
Motif organization of BsIAAs and AtIAAs proteins was investigated by MEME5.0.5 (). The maximum motif width was 50, the number of motifs was 20 [Citation20] and the other parameters were default values. The BsIAAs protein conserved domains were aligned using DNAMAN software (Version 9) ().
Figure 1. Conservative motif of B. striata(a,b) and A. thaliana(c,d).

Figure 2. Multiple sequence alignment of BsIAAs.

1.2.4 Construction of phylogenetic tree of Aux/IAA family of D. candidum and A. thaliana
Multiple sequence alignments were generated using ClustalW in MEGA 7.0 with defaulted parameters. Phylogenetic tree was generated using neighbor-joining method with 1000 bootstrap test by MEGA 7.0 software.
1.2.5 EST-SSR detection and verification
The plant samples of B. striata were collected from provinces of Sichuan, Chongqing, Guizhou and Anhui, China, for extracting genomic DNA according to the method of CTAB [Citation21]. The extracted DNA was diluted to 50 ng/μL and stored at −40°C for EST-SSR detection.
The EST-SSR markers were detected, developed and verified through the following approaches. The online software NWISRL (https://ssr.nwisrl.ars.usda.gov/) was applied to detect EST-SSR sites of each BsIAA sequence with default parameter values. Then, primers of each site were designed using DNAMAN program. Subsequently, PCR amplification was used to verify the results of PAGE detection. The PCR reaction volume was 10 ul, containing 15 ng of template DNA, 6 uL 2× PCR MIX, 0.75 uL of each primer, and 1 uL of ddH2O. The amplification conditions were: pre-denaturation at 95 °C for 5 min; denaturation at 95 °C for 30 s, annealing at 58 °C for 30 s, extension at 72 °C for 60 s, 34 cycles, and extension at 72 °C for 5 min. The amplified product was separated by a 10% polyacrylamide gel. The electrophoresis apparatus was a PowerPac type with steady-state electrophoresis apparatus. The constant voltage was set to 150 V and electrophoresis was performed for 150 min. After silver nitrate staining, the bands were observed and photographed.
2 Results
2.1 Full transcriptome data construction
After assembling the RNA-seq data of all existing Pair ends by using Trinity software, the following data were obtained (). The sequencing approach obtained 106,054,784 clean reads (SRA database accession number: SRR7058048) by using an Illumina platform, and the reads were assembled into 134,900 unigenes by the Trinity package [Citation17].
Table 1. B. striata full transcriptome data construction.
2.2 Identification of Aux/IAA gene families and analysis of protein characteristics
In this study, a total of 39 non-redundant sequences were obtained by integrating the results of the BLAST and online software Pfam verification of the transcriptome database. The resulted sequences were then searched again using Pfam batch search and 27 Aux/IAA genes with confidant domain were confirmed as representatives of BsIAA gene family after a manual curation. For the convenience of the study, the 27 BsIAAs were sequentially designated according to their sequence length, from BsIAA1 to BsIAA27 (). Information about these BsIAAs is listed in , including gene name, locus ID, ORF length, and predicted characteristics of corresponding proteins. The length of the predicted BsIAAs ranged from 1010 bp (BsIAA27) to 4335 bp (BsIAA1), with molecular weights ranging from 88.94245 kDa to 362.22076 kDa, and the deduced isoelectric points varied widely, from 4.73 (BsIAA1&2) to 5.17 (BsIAA27). The instability index analysis found that except the proteins of BsIAA3, 4, 13, 16, 17, 26, 27 were stable (unstable index < 40), the rest were unstable proteins (unstable index > 40). Subcellular localization analysis was detected to localize to the Nucleus Secondary structure analysis found that proteins of BsIAA family accounted for a large proportion of random coils, of which 26 were the largest proportion of random coils.
Table 2. Detailed information about 27 predicted Aux/IAA proteins of B. striata.
2.3 Comparative phylogenetic analysis of BsIAA
To examine the evolutionary relationships among the Aux/IAA genes from B. striata, D. officinale and A. thaliana, a rooted phylogenetic tree was generated based on the alignment of amino acid sequences for 75 Aux/IAA proteins, including 27 BsIAAs, 16 DoIAAs and 32 AtIAAs (). Phylogenetic distribution indicated that Aux/IAA proteins can be classified into two major groups (Group A and Group B), which could be further subdivided into four (A1-A4) and five (B1-B5) subgroups, respectively. Among them, group A and B consisted of 35 and 40 Aux/IAA proteins, respectively. A sister pair indicates the closest relatives within a phylogenetic tree. Within this tree, a total of 25 sister pairs were found, consisting of 10 and 15 pairs in group A and B. This pattern of two major groups for Aux/IAA gene family members in the phylogenetic tree was similar to that reported for other plants including rice [Citation22], Moso bamboo (Phyllostachys pubescens) [Citation23], soybean [Citation12] and Brassica napus [Citation2], which suggested that the Aux/IAA genes have been widely conserved in different groups.
Figure 3. Phylogeny of Aux/IAA proteins from B. striata, A. thaliana and D. officinale.
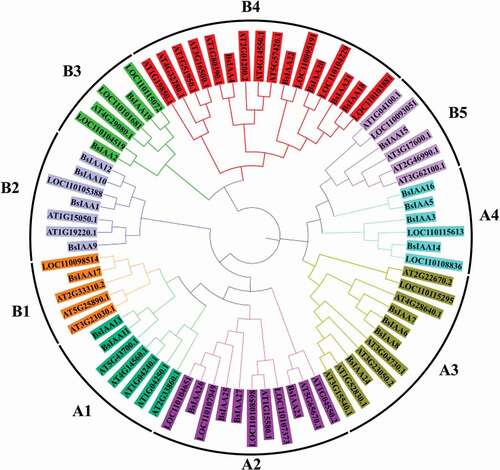
Meanwhile, to gain a better understanding of the structural diversity of the BsIAAs, we also built a separate phylogenetic tree using the same method (). Four typical domains were detected among BsIAAs, similar to the proteins of AtIAAs (). MEME analysis found that four conserved domains (Domain I to Domain IV) of BsIAAs protein were contained in five motifs (motif 1, 4, 10, 15 and 17). Except BsIAA27, the others all contained motif 1 and motif 4. Most BsIAAs did not contain motif 17, and BsIAA6, 7, 16 to 24 contained motif 10 but no Domain II. Combined with the phylogenetic tree () showed that the similar branches and types of Aux/IAA protein motifs were identical or similar to each other.
Figure 4. Phylogenetic relationships and protein motif distribution analysis.a. The phylogenetic tree of the BsIAAs. b. Motif distributions in the B. striata.
2.4 Polymorphism detection of EST-SSR in BsIAAs
In this study, 4 strains of B. striata genomic DNA were amplified with the designed 11 pairs of primers (). Among them, four pairs of primers can be amplified stably, and the length of the amplified product ranged from 70 to 300 bp (). Four DNAs amplified different polymorphic bands, and the percentage of polymorphic loci was 30%, indicating that the Aux/IAA gene family in different regions was genetically conserved and also presented different polymorphisms. Thus, SSR primers can be used as molecular markers to identify different strains of the Aux/IAA gene family.
Figure 5. SSR profiles of B. striata.
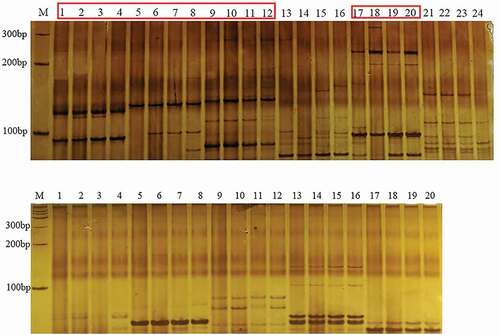
Table 3. The information of EST-SSR primers.
3 Discussion
Auxin is a key signaling molecule in the process of plant growth and development. As the earliest discovered plant hormone, its physiological role is extensive, affecting cell division, enlargement, and differentiation. Aux/IAA proteins have been suggested to bind with ARFs and prevent activation of auxin-responsive genes in the absence of auxin [Citation8]. However, in B. striata, there was very little information about the Aux/IAA genes, until total of 27 Aux/IAA genes were identified in B. striata in this paper, though lower than 29 Aux/IAA genes in A. thaliana [Citation24] and 31 Aux/IAA genes in Oryza sativa [Citation22]. However, this study was based on the transcriptome data of the B. striata. Due to incomplete data, the identified Aux/IAA genes were very few, so the whole genome could be sequenced and a database could be established in the subsequent studies.
According to the physicochemical analysis of 27 proteins of BsIAAs family, most of them acted in an acidic subcellular environment, which means they were unstable proteins. About the secondary structures, the proportion of random coils was largest in most of Aux/IAA family proteins. Subcellular localization was localized in the nucleus, suggesting that the Aux/IAA protein might play a role in the nucleus.
By analyzing the conserved domains of BsIAA gene family proteins, we found that the BsIAAs contain four domains, namely Domain I, II, III, IV. In which the Domain I at the N terminus had three repeat leucine residues, referred to as ‘LxLxL’ motif (L refers to leucine, x means any amino acid residue), which was required for the transcriptional repression function of Aux/IAA protein [Citation25], and this was the smallest and least strictly conserved among the conserved domains. Domain II contained a target site for ubiquitination degradation of Aux/IAA protein with the core sequence of VGWPP. The dominant mutation in this region made Aux/IAA protein unable to enter the ubiquitination pathway and lead to enhanced stability [Citation26]. Domain III contained a β sheet and two α helices (α1 and α2), which played an important role in the dimerization of Aux/IAA protein [Citation2]. Domain IV included the acidic region and the SV40 type NLS (PKKKRKV) [Citation26]. These were similar to A. thaliana [Citation27], Oryza sativa [Citation22], Zea mays. L [Citation11]., Cucumis sativus [Citation10] and other Aux/IAA gene family, indicating that Aux/IAA protein had a high sequence conservation. There were multiple amino acid changes in the conserved domains of BsIAA proteins. This result was similar to the analysis of the Brassica rapa Aux/IAA proteins [Citation14], indicating that the altered regions might have new functions or only some typical functions of Aux/IAA proteins.
According to the motif analysis, it indicated that these genes did not contain motif 17 might not be involved in classical auxin signal transduction. For genes containing motif 10, their protein life was longer than others [Citation12]. The results indicated that the more frequent motif was an important conserved motif in the Aux/IAA domain. Online analysis of the less frequent motif by SMART had no relevant description of its functional annotations, so it required further investigation.
Based on the phylogenetic analysis in this study, BsIAAs were divided into two classes which contained 4 and 5 subfamilies, respectively. The similar branches of Aux/IAA proteins had the same or similar motifs, such as genes between BsIAA10 and BsIAA12, BsIAA11 and BsIAA13, BsIAA18 and BsIAA20, indicating that Aux/IAA proteins were conserved, which was similar to the Aux/IAA family genes in Medicago truncatula [Citation28] and Brassica napus [Citation2]. The phylogenetic tree constructed from the Aux/IAA proteins of B. striata, D. officinale and A. thaliana showed that the B. striata Aux/IAA proteins had similar work to D. officinale, which indicated that BsIAA and DoIAAs were relatively close to each other in evolutionary relationship. This analysis could be used for further exploring the protein functions of BsIAAs.
Molecular markers are excellent tools to study the genetic relationships and genomic evolution of species. Due to the conserved nature of flanking sequences, SSR markers developed in one species can be employed to detect these microsatellite loci in other related species [Citation29]. In this study, EST-SSR site analysis was performed on BsIAAs, revealing that the ratio of polymorphic alleles was 30%, which means B. striata in different regions have high genetic conservatism. And it was mainly due to the conservation of Aux/IAA gene itself, but it also showed the genetic diversity. Therefore, SSR molecular markers could be used to evaluate the genetic diversity in different regions. We identified the genetic relationship by using the amplified primers in different regions and number of bands, and estimate the genetic relationship in order to provide more accurate information for breeding, breed identification and genetic structure research.
Auxin plays a very important role in plants, affecting the yield and quality of B. striata. Based on the first transcriptome databases of B. striata, we identified the 27 members of the Aux/IAA gene family and analyzed their basic physicochemical properties, subcellular localization, protein conserved domains, conserved motifs and phylogenetics in this paper. Transcriptome gene expression characteristics of the group were comprehensively analyzed and SSR molecular markers were also performed. In conclusion, these genes can be divided into group A and group B, which contain 4 and 5 subfamilies, respectively. Among them, BsIAAs were more closer to those of D. officinale compared to A. thaliana. In this study, a total of 11 pairs of primers were designed, among which 4 pairs could be amplified stably and showed different polymorphism. These results laid a solid foundation for further study of the biological functions of BsIAAs, as well as the identification of B. striata germplasm resources and the analysis of phylogenetic relationships.
Ethics Statement
The Bletilla striata seeds used in this study were purchased from Menghe Ran, a farmer planting medicinal herbs in Zheng’an County (28°56′N, 107°43′E), Guizhou Province of China, on October 20th of 2015 with the price of 10 RMB per capsule. Then, the seeds were only tissue cultured in labs of Chinese Medical Herb Research Group on main campus of Zunyi Medical University locating in Xinpu District, Zunyi City, Guizhou Province of China. All of the experiments conducted on the seeds, seedlings and plants by the research group were thoroughly comply by the requirements on plant researches of common ethics and the rules of the university.
The B. striata plants for harvesting capsules were first collected by Ran’s family from Zheng’an areas many years ago. The farmer who sold materials to the research group completely agreed all of the researches on the materials, including landraces’ capsule seeds, plants and tubers bought from him and hoping the researchers can get improvement for helping his business.
Interest statement
The authors declare that they have no conflict of interest.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Yang F, Song Y, Yang H, et al. An auxin-responsive endogenous peptide regulates root development in Arabidopsis. J Integr Plant Biol. 2014;56(7):635–647.
- Li H, Wang B, Zhang Q, et al. Genome-wide analysis of the auxin/indoleacetic acid (Aux/IAA) gene family in allotetraploid rapeseed (Brassica napus L.). BMC Plant Biol. 2017;17(1):204–213.
- Kepinski S, Leyser O. Auxin-induced SCFTIR1-Aux/IAA interaction involves stable modification of the SCFTIR1 complex. Proc Nat Acad Sci. 2004;101(33):12381–12386.
- Hu W, Zuo J, Hou X, et al. The auxin response factor gene family in banana: genome-wide identification and expression analyses during development, ripening, and abiotic stress. Front Plant Sci. 2015;6(undefined):742–757.
- Hayashi K. The interaction and integration of auxin signaling components. Plant Cell Physiol. 2012;53(6):965–975.
- Li F, Wu M, Liu H, et al. Systematic identification and expression pattern analysis of the Aux/IAA and ARF gene families in moso bamboo (Phyllostachys edulis). Plant Physiol Biochem. 2018;130:431–444.
- Rybel BD, Vassileva V, Parizot B, et al. A novel Aux/IAA28 signaling cascade activates GATA23-dependent specification of lateral root founder cell identity. Curr Biol. 2010;20(19):1697–1706.
- Luo J, Zhou JJ, Zhang JZ. Aux/IAA gene family in plants: molecular structure, regulation, and function. Int J Mol Sci. 2018;19(1):259–274.
- Yu H, Soler M, San CH, et al. Comprehensive genome-wide analysis of the Aux/IAA gene family in Eucalyptus: evidence for the role of EgrIAA4 in wood formation. Plant Cell Physiol. 2015;56(4):700–714.
- Gan D, Zhuang D, Ding F, et al. Identification and expression analysis of primary auxin-responsive Aux/IAA gene family in cucumber (Cucumis sativus). J Genet. 2013;92(3):513–521.
- Wang Y, Deng D, Bian Y, et al. Genome-wide analysis of primary auxin-responsive Aux/IAA gene family in maize (Zea mays. L.). Plos One. 2010;37(8):3991–4001.
- Singh VK, Jain M. Genome-wide survey and comprehensive expression profiling of Aux/IAA gene family in chickpea and soybean. Front Plant Sci. 2015;6:918–932.
- Ke Y, Abbas F, Zhou Y, et al. Genome-wide analysis and characterization of the Aux/IAA Family genes related to floral scent formation in hedychium coronarium. Int J Mol Sci. 2019;20(13):3235–3255.
- Paul P, Dhandapani V, Rameneni JJ, et al. Genome-wide analysis and characterization of Aux/IAA family Genes in Brassica rapa. PLoS One. 2016;11(4):e0151522.
- Lingna C, Xianggan Z, Xiaojuan G, et al. The roles of Aux/IAA gene family in development of Dendrocalamus sinicus (Poaceae: bambusoideae) inferred by comprehensive analysis and expression profiling. Mol Biol Rep. 2019;46:1625–1634.
- Chen Z, Cheng L, He Y, et al. Extraction, characterization, utilization as wound dressing and drug delivery of Bletilla striata polysaccharide: A review. Int J Biol Macromol. 2018;120:2076–2085.
- Xu DL, Chen HB, Aci M, et al. De Novo assembly, characterization and development of EST-SSRs from Bletilla striata transcriptomes profiled throughout the whole growing period. PLoS One. 2018;13(10):e0205954.
- Kumar R, Tyagi AK, Sharma AK, et al. Genome-wide analysis of auxin response factor (ARF) gene family from tomato and analysis of their role in flower and fruit development. Mol Genet Genomic. 2011;285(3):245–260.
- Huang X, Bao YN, Wang B, et al. Identification and expression of Aux/IAA, ARF, and LBD family transcription factors in Boehmeria nivea. BMC Plant Biol. 2016;60(2):244–250.
- Liu H, Wu M, Zhu D, et al. Genome-Wide analysis of the AAAP gene family in moso bamboo (Phyllostachys edulis). BMC Plant Biol. 2017;17(1):29–46.
- Aboul-Maaty NAF, Oraby HAS. Extraction of high-quality genomic DNA from different plant orders applying a modified CTAB-based method. Bull National Res Centre. 2019;43(1). DOI:https://doi.org/10.1186/s42269-019-0066-1
- Jain M, Kaur N, Garg R, et al. Structure and expression analysis of early auxin-responsive Aux/IAA gene family in rice (Oryza sativa). Funct Integr Genomics. 2006;6(1):47–59.
- Wang W, Gu L, Ye S, et al. Genome-wide analysis and transcriptomic profiling of the auxin biosynthesis, transport and signaling family genes in moso bamboo (Phyllostachys heterocycla). BMC Genomics. 2017;18(1):870–885.
- Fattorini L, Ronzan M, Piacentini D, et al. Cadmium and arsenic affect quiescent centre formation and maintenance in Arabidopsis thaliana post-embryonic roots disrupting auxin biosynthesis and transport. Environ Exp Bot. 2017;144:37–48.
- Li H, Tiwari SB, Hagen G, et al. Identical amino acid substitutions in the repression domain of auxin/indole-3-acetic acid proteins have contrasting effects on auxin signaling. Plant Physiol. 2011;155(3):1252–1263.
- Guilfoyle TJ, Hagen GJ. Getting a grasp on domain III/IV responsible for auxin response factor–IAA protein interactions. Plant Sci. 2012;190(3):82–88.
- Carrier DJ, Bakar NT, Swarup R, et al. The binding of auxin to the Arabidopsis auxin influx transporter AUX1. Plant Physiol. 2008;148(1):529–535.
- Shen C, Yue R, Yang Y, et al. Genome-wide identification and expression profiling analysis of the Aux/IAA gene family in Medicago truncatula during the early phase of Sinorhizobium meliloti infection. PLoS One. 2014;9(9):e107495.
- Thakur AK, Singh KH, Singh L, et al. SSR marker variations in Brassica species provide insight into the origin and evolution of Brassica amphidiploids. Hereditas. 2018;155(6). DOI:https://doi.org/10.1186/s41065-017-0041-5