

ABSTRACT
Work undertaken using the embryonic carcinoma 2102Ep line, highlighted the requirement for robust, well-characterized and standardized protocols. A systematic approach utilizing ‘quick hit’ experiments demonstrated variability introduced into culture systems resulting from slight changes to culture conditions (route A). This formed the basis for longitudinal experiments investigating long-term effects of culture parameters including seeding density and feeding regime (route B).Results demonstrated that specific growth rates (SGR) of passage 59 (P59) cells seeded at 20,000 cells/cm2 and subjected to medium exchange after 48h prior to reseeding at 72h (route B2) on average was marginally higher than, P55 cells cultured under equivalent conditions (route A1); whereby SGR values were (0.021±0.004) and (0.019±0.004). Viability was higher in route B2 over 10 passages with average viability reported as (86.3%±8.1) compared to route A1 (83.3±8.8). The metabolite data demonstrated both culture route B1 (P57 cells seeded at 66,667 cells/cm2) and B2 had consistent-specific metabolite rates (SMR) for glucose, but SMR values of route B1 was consistently lower than route B2 (0.00001 mmol, cell-1.d-1 and 0.000025).Results revealed interactions between phenotype, SMR and feeding regime that may not be accurately reflected by growth rate or observed morphology. This implies that current schemes of protocol control do not adequately account for variability, since key cell characteristics, including phenotype and SMR, change regardless of standardized seeding densities. This highlights the need to control culture parameters through defined protocols, for processes that involve culture for therapeutic use, biologics production, and reference lines.
GRAPHICAL ABSTARCT
Research Highlights
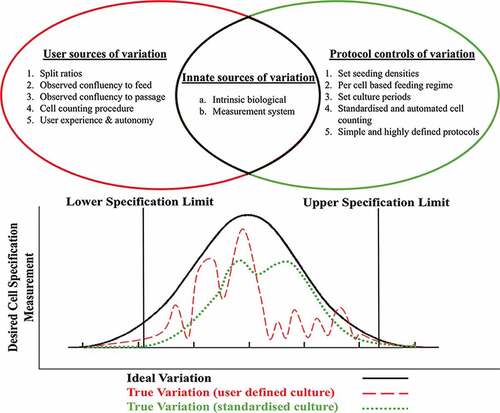
Subtle, often unintentional variations in culture parameters have a significant impact on cultured cells.
The importance of balancing key nutrients, gene expression, phenotype and feeding regimes was shown.
Standardization is crucial to ensure manufacturing process reproducibility and comparability.
This is integral to the pragmatic and successful use of reference lines in cell therapy manufacturing.
Our experimental methodical approach demonstrates the complex interactions and balance required between key nutrients, gene expression, cell phenotype and feeding regimes employed that cannot be accurately represented by growth rate and cell counts alone.
Standardised protocols are crucial not only from a uniformity perspective, but also for to ensure process reproducibility and comparability, particularly in the decentralised manufacturing of cell-based products.
This is integral to the pragmatic and successful use of reference cell lines in cell therapy manufacturing.
Acknowledgements
The authors acknowledge the National Institute for Biological Standards and Control (NIBSC).
Author Contribution Statements
James Willard Tonderai Kusena, Maryam Shariatzadeh, Adam James Studd, Jenna Rebekah James, Robert James Thomas and Samantha Louise Wilson have provided substantial contributions to the manuscript. JWTK, RJT and SLW were involved in the conception or design of the work; JWTR, MS, AJS, JRJ, RJT and SLW were involved with acquisition, analysis, or interpretation of data for the work; AND all authors were involved with drafting the work or revising it critically for important intellectual content. All authors give their final approval for the submission and publication of this manuscript in Bioengineered and agree to be accountable for all aspects of this work.
Disclosure statement
James Willard Tonderai Kusena, Maryam Shariatzadeh, Adam James Studd, Jenna James, Robert James Thomas and Samantha Louise Wilson have no conflict of interest to disclose.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Supplementary Material
Supplemental data for this article can be accessed here.