

ABSTRACT
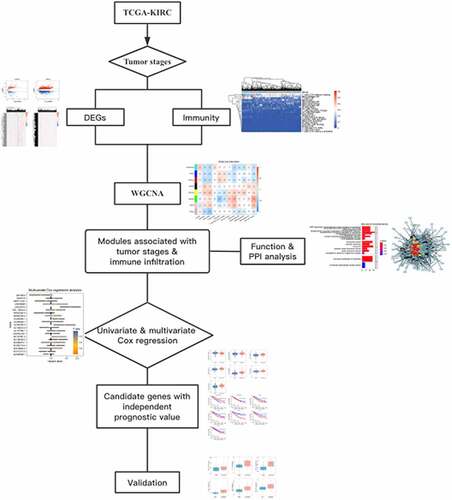
The clear cell renal cell carcinoma (ccRCC) is the main pathological subtype of renal cell carcinoma. Immune system evasion, one hallmark of cancer, contributes to cancer cells in escaping from the attack of immune cells. In order to identify potential prognostic biomarkers in ccRCC patients and immune cells fraction, we collected and downloaded profiles from The Cancer Genome Atlas (TCGA) database and Gene Expression Omnibus (GEO) database. We obtained 2 modules significantly associated with tumor stage and immune cells; functional enrichment analysis showed that genes in the module ‘yellow’ were significantly enriched in proteins targeting to membrane and ribosome, as well as the oxidative phosphorylation pathway, while genes in the module ‘green’ mainly participate in molecular functions associated with immunity like activation of T cells. Four LncRNAs (LINC00472, AL590094.1, AL365203.3, and AC147651.3) and RPL27A and RPL22L1 in the module ‘yellow’ and two lncRNAs (LINC00426 and AC129507.2) and five protein-coding genes (CSF1, NOD2, ITGAE, CD7, and PDCD1) in the module ‘green’ represented independent prognostic values in patients with ccRCC. Expression of LINC0042, NOD2, CD7, and PDCD1 were significantly correlated with ratio of immune cells (like T cells CD8 and resting mast cells). LINC00426, with significant correlation with immune cell fraction, shows potential prognostic value in ccRCC patients. Our findings provide a strategy in exploring biomarkers with prognostic significance and significant association with the fraction of immune cells.
GA
Highlights
Six lncRNAs (LINC00472, AL590094.1, AL365203.3, AC147651.3, AC129507.2, and LINC00426) and 7 genes (RPL27A, RPL22L1, CSF1, NOD2, ITGAE, CD7, and PDCD1) show expression differences in patients with KIRC at early stages compared to those at advanced stages;
Six lncRNAs and 7 genes represent independent prognostic significance.
LINC00426 is one potential biomarker of prognostic value and indicates a significant correlation with immune cell fraction.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Availability of data and material
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author contributions
Zhenfei Xiang: Data curation, Writing-Original draft preparation.
Erdong Shen: Visualization, Investigation.
Mingyao Li: Conceptualization, Methodology.
Danfei Hu: Software.
Zhanchun Zhang: Validation
Senquan Yu: Writing- Reviewing and Editing, Supervision.
Supplementary material
Supplemental data for this article can be accessed here.