

ABSTRACT
Breast cancer is the most commonly diagnosed malignancy in women; thus, more cancer prevention research is urgently needed. The aim of this study was to predict potential therapeutic agents for breast cancer and determine their molecular mechanisms using integrated bioinformatics. Summary data from a large genome-wide association study of breast cancer was derived from the UK Biobank. The gene expression profile of breast cancer was from the Oncomine database. We performed a network-wide association study and gene set enrichment analysis to identify the significant genes in breast cancer. Then, we performed Gene Ontology analysis using the STRING database and conducted Kyoto Encyclopedia of Genes and Genomes pathway analysis using Cytoscape software. We verified our results using the Gene Expression Profile Interactive Analysis, PROgeneV2, and Human Protein Atlas databases. Connectivity map analysis was used to identify small-molecule compounds that are potential therapeutic agents for breast cancer. We identified 10 significant genes in breast cancer based on the gene expression profile and genome-wide association study. A total of 65 small-molecule compounds were found to be potential therapeutic agents for breast cancer.
Graphical Abstract
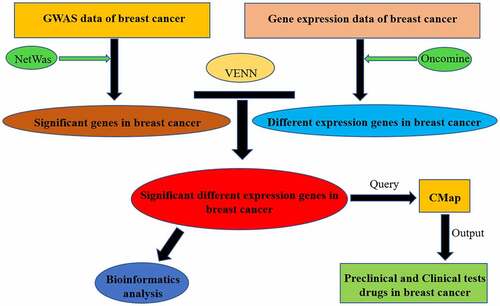
Acknowledgements
The authors acknowledge the National Natural Science Foundation of China 81972861, Innovation Capacity Support Plan of Shaanxi Province 2018TD‐002. We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Availability of data and materials
All materials are available by the corresponding author.
Declarations
Ethical statement
Our study did not require an ethical board approval because it did not contain human or animal trials.
Consent for publication
Not applicable.
Disclosure Statement
The authors declare no competing interests.
Highlights
Combined analyses of network-wide association studies, gene expression profiles, and drug databases.
A useful approach for evaluating the relationship among genes, diseases, and drugs.
These findings will pave the way for the discovery of potential therapeutic targets for breast cancer.
Supplementary Material
Supplemental data for this article can be accessed here.