

ABSTRACT
Oral squamous cell carcinoma (OSCC) is a common human malignancy. However, its pathogenesis and prognostic information are poorly elucidated. In the present study, we aimed to probe the most significant differentially expressed genes (DEGs) and their prognostic performance in OSCC. Multiple microarray datasets from the Gene Expression Omnibus (GEO) database were aggregated to identify DEGs between OSCC tissue and control tissue. Least absolute shrinkage and selection operator (LASSO) Cox model was constructed to determine the prognostic performance of the aggregated DEGs based on The Cancer Genome Atlas (TCGA) OSCC cohort. Ten datasets with 341 OSCC samples and 283 control samples were included. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment revealed that the integrated DEGs were enriched in the IL-17 signaling pathway, viral protein interactions with cytokines and cytokine receptors, and amoebiasis, among others. Our LASSO Cox model was able to discriminate two groups with different overall survival in the training cohort and test cohort (p < 0.001). The time-dependent receiver operating characteristic (ROC) curve revealed that the area under the curve (AUC) values at one year, three years, and five years were 0.831, 0.898, and 0.887, respectively. In the testing cohort, the time-dependent ROC curve showed that the AUC values at one year, three years, and five years were 0.696, 0.693, and 0.860, respectively. Our study showed that the integrated DEGs of OSCC might be applicable in the evaluation of prognosis in OSCC. However, further research should be performed to validate our findings.
Graphical abstract
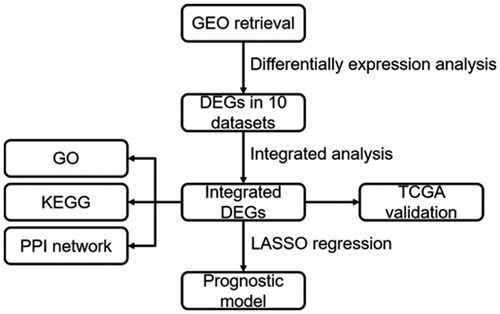
Highlights
MMP1, MMP10, MMP3, MMP13, and MMP12 were the most highly upregulated genes in OSCC.
CRISP3, MAL, KRT4, TMPRSS11B, and CRNN were the most highly downregulated genes in OSCC.
The differentially expressed genes of OSCC might be applicable in the evaluation of prognosis in OSCC.
Acknowledgements
We would like to thank TCGA project and all researchers contributing to GEO datasets.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Ethics approval and consent to participate
Not applicable. All data was derived from TCGA and GEO database, and we obeyed the usage of principle of TCGA and GEO.
Authors’ contributions
YZ: Methodology, Investigation, Software, Validation, Data curation, Validation, Formal analysis, Writing - original draft. JH: Methodology, Investigation, Validation, Data curation, Validation, Formal analysis, Writing - original draft. JC: Conceptualization, Supervision, Writing - review & editing. All authors read and approved the final manuscript.
Consent for publication
Not applicable. Individual information involved in this study was derived from public database (TCGA and GEO).
Data availability statement
The datasets analyzed was acquired from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/) and GEO database (https://www.ncbi.nlm.nih.gov/geo/).
Supplementary material
Supplemental data for this article can be accessed here.