

ABSTRACT
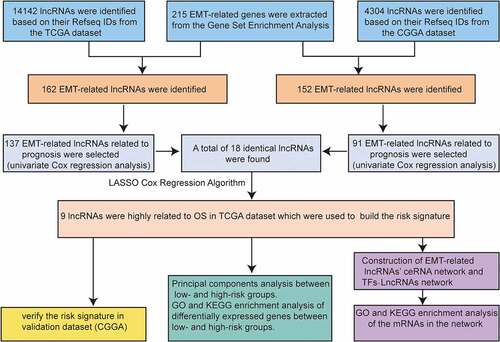
Epithelial–mesenchymal transition (EMT)-related long non-coding RNAs (lncRNAs) may be exploited as potential therapeutic targets in gliomas. However, the prognostic value of EMT-related lncRNAs in gliomas is unclear. We obtained lncRNAs from The Cancer Genome Atlas and constructed EMT-related lncRNA co-expression networks to identify EMT-related lncRNAs. The Chinese Glioma Genome Atlas (CGGA) was used for validation. Gene set enrichment and principal component analyses were used for functional annotation. The EMT–lncRNA co-expression networks were constructed. A real-time quantitative polymerase chain reaction assay was performed to validate the bioinformatics results. A nine-EMT-related lncRNAs (HAR1A, LINC00641, LINC00900, MIR210HG, MIR22HG, PVT1, SLC25A21-AS1, SNAI3-AS1, and SNHG18) signature was identified in patients with glioma. Patients in the low-risk group had a longer overall survival (OS) than those in the high-risk group (P < 0.0001). Additionally, patients in the high-risk group showed no deletion of chromosomal arms 1p and/or 19q, isocitrate dehydrogenase wild type, and higher World Health Organization grade. Moreover, the signature was identified as an independent factor and was significantly associated with OS (P = 0.041, hazard ratio = 1.806). These findings were further validated using the CGGA dataset. The low- and high-risk groups showed different EMT statuses based on principal component analysis. To study the regulatory function of lncRNAs, a lncRNA-mediated ceRNA network was constructed, which showed that complex interactions of lncRNA–miRNA–mRNA may be a potential cause of EMT progression in gliomas. This study showed that the nine-EMT-related lncRNA signature has a prognostic value in gliomas.
Graphical Abstract
Acknowledgements
The authors gratefully acknowledge contributions of the CGGA network and the TCGA Network. And we acknowledge Luyue Chen for his contribution in the revision of the article.
Disclosure statement
The authors declare that they have no competing interests.
Author contribution
Tao CM and Luo HT performed the data analysis work and aided in writing the manuscript. Li JY collected and processed the data. Tao CM and Luo HT performed the qRT-PCR experiments. Chen LY, Huang K and Zhu XG edited the manuscript. All authors read and approved the final manuscript.
Supplementary material
Supplemental data for this article can be accessed here