

ABSTRACT
Rectal cancer is a life‑threatening disease worldwide. Chemotherapy resistance is common in rectal adenocarcinoma patients and has unfavorable survival outcomes; however, its related molecular mechanisms remain unknown. To identify genes related to the initiation and progression of rectal adenocarcinoma, three datasets were obtained from the Gene Expression Omnibus database. In total, differentially expressed genes were analyzed from 294 tumor and 277 para-carcinoma samples from patients with rectal cancer. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes functions were investigated. Cytoscape software and MicroRNA Enrichment Turned Network were applied to construct a protein-protein interaction network of the dependent hub genes and related microRNAs. The Oncomine database was used to identify hub genes. Additionally, Gene Expression Profiling Interactive Analysis was applied to determine the RNA expression level. Tumor immune infiltration was assessed using the Tumor Immune Estimation Resource database. The expression profiles of hub genes between stages, and their prognostic value, were also evaluated. During this study, data from The Cancer Genome Atlas were utilized. In rectal adenocarcinoma, four hub genes including CXCL1, CXCL2, CXCL3, and GNG4 were highly expressed at the gene and RNA levels. The expression of CXCL1, CXCL2, and CXCL3 was regulated by has-miR-1-3p and had a strong positive correlation with macrophage and neutrophil. CXCL2 and CXCL3 were differentially expressed at different tumor stages. High expression levels of CXCL1 and CXCL3 predicted poor survival. In conclusion, the CXCL1 and CXCL3 genes may have potential for prognosis and molecular targeted therapy of rectal adenocarcinoma.
Graphical abstract
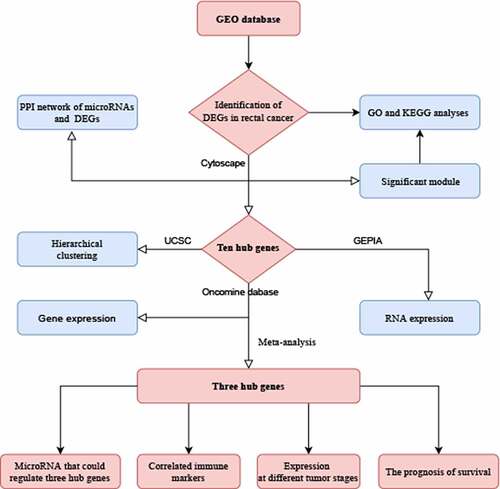
Research highlights
In READ, the expression of CXCL1, CXCL2, and CXCL3 had a strong positive correlation with macrophage and neutrophil.
Gene CXCL2 and CXCL3 had various expression levels in READ patients at different stages.
Overexpression of CXCL1 and CXCL3 were found to have prognostic values with READ.
Acknowledgements
The authors thank all the patients who participated in this study and their families and the technical support from Omigen, Inc.
Disclosure statement
No potential conflict of interest was reported by the authors.
Ethics approval and consent to participate
There was no ethics associated with this work because the study did not involve animal experiments or human specimens.
Data availability statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors’ contributions
CLZ and MS conceived and designed the study with QYL and HZZ. QYL and HZZ drafted the manuscript and analyzed the data with YYX and ZYS. RQW, KJL, XD, and DNG formatted the images and the article. HXJ reviewed the data. All authors read and approved the final manuscript.