

ABSTRACT
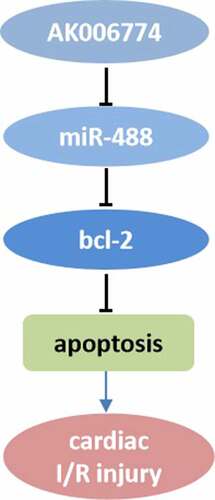
In recent years, the incidence and mortality of myocardial infarction (MI) have been increasing throughout the world, threatening public health. Non-coding RNAs (ncRNAs), including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), play critical roles in the progression of MI. The present study aimed to investigate the role of lncRNA AK006774 in the progression of myocardial infarction and find out novel therapeutic or diagnostic target of myocardial infarction. A mouse ischemia/reperfusion (I/R) model and 2,3,5-Triphenyte-trazoliumchloride (TTC) staining were performed to evaluate the effects of AK006774 on I/R injury in vivo. Hypoxia/reoxygenation (H/R) models using primary cardiomyocytes have been established. Flow cytometry and Terminal Deoxynucleotide Transferase dUTP Nick End Labeling (TUNEL) assays were performed to evaluate the effects of AK006774 on cardiomyocyte apoptosis. Luciferase and RNA pull-down assays were performed to verify the interaction between miR-448 and its targets. Western blotting and quantitative PCR were performed to determine protein and gene expression, respectively. We first found that AK006774 overexpression reduced I/R-induced infarct area and cardiomyocyte apoptosis in vivo. Accordingly, AK006774 inhibited apoptosis and oxidative stress in cardiomyocytes subjected to H/R treatment in vitro. Mechanistically, AK006774 modulated the expression of bcl-2 by sponging miR-448. Overexpression of miR-448 antagonized the effects of AK006774 on cardiomyocyte apoptosis. The AK006774/miR-448/bcl-2 signaling axis acts as a key regulator of I/R injury and may be a potential therapeutic or diagnostic target for the treatment of MI.
Graphical abstract
Highlights
Silencing of lncRNA AK006774 which is upregulated in cardiac ischemia reperfusion can alleviate the I/R injury
AK006774 sponges miR-488 in cardiomyocytes
miR-448 targets bcl-2 in cardiomyocytes
Disclosure statement
No potential conflict of interest was reported by the author(s).