

ABSTRACT
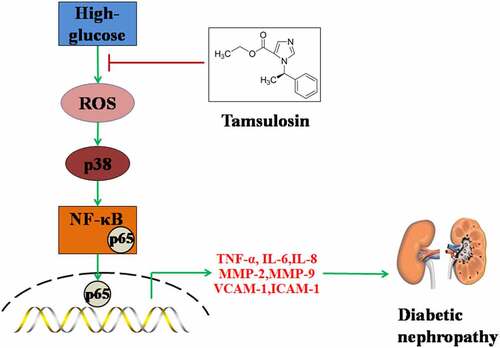
Diabetic nephropathy (DN) is a common complication of diabetes. Tamsulosin is a selective α1-AR antagonist. α1-AR is expressed widely in kidney tissues and has displayed its various physiological functions. However, whether Tamsulosin has affects DN is unknown. To our knowledge, this is the first time it has been examined whether Tamsulosin possesses a beneficial effect in high glucose-challenged glomerular endothelial cells (GECs). Firstly, we found that Tamsulosin reduced high glucose-induced expressions of TNF-α, IL-6, and IL-8. Secondly, Tamsulosin alleviated high glucose-induced expressions of MMP-2 and MMP-9. Thirdly, Tamsulosin inhibited the expressions of VCAM-1 and ICAM-1. Importantly, our results indicate that Tamsulosin inhibited high glucose-induced expressions of fibrosis factors such as Col-1 and TGF-β1. Additionally, we found that Tamsulosin ameliorated oxidative stress via reducing the generation of ROS and preventing the activation of p38. Mechanistically, we found that Tamsulosin attenuated high glucose-induced activation of NF-κB. Based on these findings, we conclude that Tamsulosin could attenuate high glucose-induced injury in GECs through alleviating oxidative stress and inflammatory response.
Graphical abstract
Acknowledgements
This study is funded by the “Observation of Therapeutic Effect of Shenlingbaizhu Granule Combined with Abelmoschus Manihot Membranous Nephropathy in Rats (2020-085)” and the “Natural Science Foundation of Heilongjiang province (LH2020H061)”.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here