

ABSTRACT
Clear cell renal cell carcinoma (ccRCC) is the most common subtype of renal cancer. Currently, we lack effective risk models for the prognosis of ccRCC patients. Given the significant role of cancer immunity in ccRCC, we aimed to establish a novel united risk model including clinical stage and immune-related gene pairs (IRGPs) to assess the prognosis. The gene expression profile and clinical data of ccRCC patients from The Cancer Genome Atlas and Arrayexpress were divided into training cohort (n = 381), validation cohort 1 (n = 156), and validation cohort 2 (n = 101). Through univariate Cox regression analysis and Least Absolute Shrinkage and Selection Operator analysis, 11 IRGPs were obtained. After further analysis, it was found that clinical stage could be an independent prognostic factor; hence, we used it to construct a united prognostic model with 11 IRGPs. Based on this model, patients were divided into high-risk and low-risk groups. In Kaplan–Meier analysis, a significant difference was observed in overall survival (OS) among all three cohorts (p < 0.001). The calibration curve revealed that the signature model is in high accordance with the observed values of each data cohort. The 1-year, 3-year, and 5-year receiver operating characteristic curves of each data cohort showed better performance than only IRGP signatures. The results of immune infiltration analysis revealed significantly (p < 0.05) higher abundance of macrophages M0, T follicular helper cells, and other tumor infiltrating cells. In summary, we successfully established a united prognostic risk model, which can effectively assess the OS of ccRCC patients.
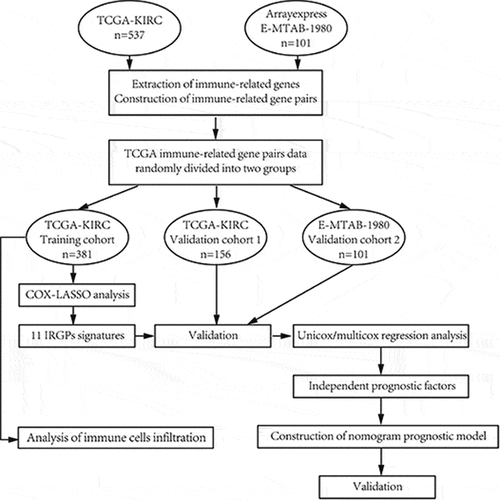
Graphical Abstract
Research highlights
·Paired comparison of immune gene expression values effectively reduces platform bias.
·Immune-related gene pairs were combined with stage to better predict the prognosis.
·Immune cell infiltration plays a role in the occurrence and development of tumors.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data accessibility
The expression and clinical information data used to support the findings of this study have been deposited in the TCGA (https://portal.gdc.cancer.gov/) and Arrayexpress(https://www.ebi.ac.uk/arrayexpress/) repository. The immune-related genes list was achieved from the Immport Shared Gene Lists Data(https://www.immport.org/home). The data of all the above databases can be downloaded freely, and our research complied with the agreement of these databases.
Author contributions
Zijia Tao: Conceptualization, Methodology, Software; Enchong Zhang: Validation, Formal analysis; Lei Li: Resources, Data Curation; Jianyi Zheng: Writing – Original Draft, Writing – Review & Editing; Yiqiao Zhao: Visualization; Xiaonan Chen: Funding acquisition, Project administration, Supervision