

ABSTRACT
Tumor-infiltrating immune cells (TICs) can serve as an important indicator to evaluate the prognosis and therapeutic response in lung adenocarcinoma (LUAD). The identification of mutated genes that can affect the abundance of TICs and prognosis has practical implications. In the presented study, tumor microenvironment (TME) scoring was performed by the ESTIMATE scoring system on 598 RNA transcripts selected from the TCGA database to determine the proportions of immune cells and stromal cells. The infiltration difference of TICs in LUAD samples was obtained by CIBERSORT. The ‘immuneeconv’ R software package, which integrates six latest algorithms, including TIMER, xCell, MCP-counter, CIBERSORT, EPIC and quanTIseq were used to verify the correlation between purinergic receptor P2Y13 (P2RY13) and immune cells. Based on RNA sequencing analysis of the Lewis lung cancer-bearing model in C57BL/6 mice and immunohistochemistry (IHC) of human LUAD tissues, the expression of P2RY13 and associated pathways were verified. It was shown that differentially expressed genes (DEGs) obtained by interactive analysis based on Immunescore and Stromalscore were significantly enriched in immune-related pathways. The expression of P2RY13 was significantly associated with prognosis and clinicopathological characteristics of LUAD patients. More importantly, this gene played an important role in maintaining the immune dominant environment and changing the regulation of TICs. P2RY13 expression was positively correlated with the infiltration of dendritic cells (DCs) in various of tumor tissues as validated by the PanglaoDB scRNA-seq database. Therefore, P2RY13 is expected to be a potential biomarker for predicting TME and the prognosis of LUAD after verification.
GRAPHICAL ABSTRACT
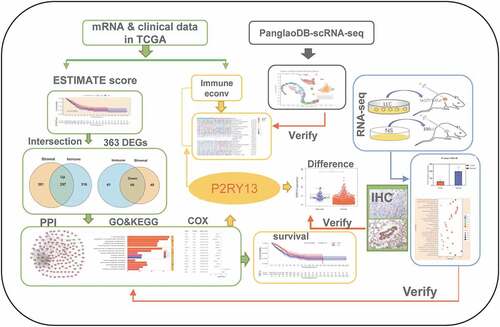
Highlights
P2RY13 was associated with prognosis and clinicopathological features in LUAD.
P2RY13 was enriched in immune-related pathways and down-regulated in LUAD.
P2RY13 was associated with infiltration of major immune cells especially DCs.
Acknowledgements
The authors would like to express their gratitude to American Journal Experts (AJE) (https://secure.aje.com/en/certificate) for the expert linguistic services provided.
Disclosure statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Authorship contribution statement
Jiangtao Wang, Weiwei Shi, Juntao Ran: Conceptualization, Methodology, Software, Formal analysis, Validation, Experiments, Data curation, Writing - original draft, Writing - review & editing, Visualization. Jiangtao Wang, Yandong Miao: Methodology, Software, Formal analysis, Writing-original draft. Jian Gan, Jiangtao Wang: Methodology, Validation, Formal analysis, Investigation. Jiangtao Wang, Jian Gan: Data curation, Investigation. Juntao Ran, Quanlin Guan: Conceptualization, Writing-review & editing, Supervision, Project administration.
Availability of data and materials
Please refer to the raw RNA-seq data of C57BL/6 mouse Lewis tumor-bearing model that we uploaded to BioSample database through the following link: https://www.ncbi.nlm.nih.gov/sra/PRJNA752479. For additional raw data, please contact the corresponding author for more information.
Ethics approval and consent to participate
This study was approved by the Institutional Review Board of the First Hospital of Lanzhou University (LDYYLL2019-130). The Ethics Committee of the First Hospital of Lanzhou University has agreed to avoid signing informed consent when obtaining wax blocks of human lung adenocarcinoma tissue.
Supplementary material
supplemental data for this article can be accessed here.