
ABSTRACT
Alcoholic liver disease (ALD) has seriously harmed the health of people worldwide, but its underlying mechanisms remain unclear. This study aims to clarify the biological function of microRNA-378b (miR-378b) in ethanol (EtOH)-induced hepatic lipid accumulation. Here, we report miR-378b is over-expressed in EtOH-induced cells and EtOH-fed mice and finally accelerates lipid accumulation. MiR-378b directly targets Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2), a kinase of AMP-activated protein kinase (AMPK), and mediates the protein level of CaMKK2. Over-expression of miR-378b exacerbated the lipid accumulation induced by EtOH and inhibited CaMKK2 and the AMPK cascade while inhibition of miR-378b ameliorated lipid metabolism dysfunction in vivo and in vitro. In brief, our results show that miR-378b plays an important role in the regulation of lipid metabolism by directly targeting CaMKK2.
1. Introduction
The misuse of alcohol is estimated to be one of the world’s largest risk factors and the main cause of several diseases, especially ALD [Citation1]. For example, the prevalence of ALD is about 2% in the general population in the United States. In some areas of China, the prevalence has been reported to range from 2.3% to 6.1% of the the total population. In addition, 85–90% alcohol-abusers develop ALD, which finally accounts for 4% of mortality worldwide [Citation2,Citation3]. The clinical symptoms of ALD vary, and ALD can progress from reversible hepatic steatosis, steatohepatitis, and fibrosis to cirrhosis and rarely to eventual hepatocellular carcinoma [Citation4]. Without treatment, hepatic steatosis, the earliest stage of ALD, may develop into the advanced stages of ALD [Citation5]. It is believed that intemperants which chronically consume alcohol heavily or consistently develop steatosis partly due to dysfunction of lipid metabolism caused by grown de novo lipogenesis and lessened fatty acid oxidation [Citation6]. Although great efforts have been made to improve our understanding of ALD, available therapies for ALD are limited. Alcohol abstinence and relapse prevention are still the basis of treatment for ALD [Citation7]. Therefore, the need to identify an efficient therapeutic target in primary ALD is urgent.
CaMKK2, one of the most versatile CaMKs, is a powerful regulator of energetic balance in the body that phosphorylates and activates AMPK to mediate many essential physiological and pathophysiological processe [Citation8]. CaMKK2 serves as a direct upstream kinase of AMPK, a key regulator of energetic balance both at the cellular level and in the whole body, and CaMKK2 can bind AMPK, forming a special signaling complex [Citation9]. AMPK is a classic node that regulates lipid metabolism processes by directly affecting protein expression or modification, as well as the transcription of lipid metabolism-related factors [Citation10]. For instance, AMPK monitors overall lipid metabolism by directly phosphorylating acetyl-CoA carboxylase (ACC) which stabilizes lipid levels by regulating downstream factors related to the synthesis and decomposition of fatty acids [Citation11]. Beyond that, AMPK suppresses transcription factors that activate glycolytic and lipogenic transcriptional programs, most notably sterol regulatory element binding protein 1 (SREBP1), which preferentially regulates the lipogenic process by activating genes involved in fatty acid and triglyceride (TG) synthesis [Citation12]. Research has shown that M3 muscarinic receptor induces AMPK phosphorylation by activating the CaMKK2 and ultimately decreases hepatocyte lipid accumulations [Citation13]. Growing evidence has revealed the significance of CaMKK2 in regulating hepatic lipid metabolism via the AMPK pathway and suggests that regulatory function of CaMKK2 may play a major part in hepatic lipid metabolic. Due to these findings, it is important to clarify whether CaMKK2 can relieve EtOH-induced hepatic steatosis by regulating hepatic lipid metabolism.
Human miR-378b, a member of the miR-378 family, is a small non-coding RNA (~22 nucleotides) that is highly conserved across species. Previous studies reported that abnormal expression of hepatic miR-378/378* was confirmed in rat models of type 2 diabetes and obese [Citation14]. MiR-378* up-regulation in breast cancer promoted lactic acid production and reduced the tricarboxylic acid cycle and oxygen consumption, suggesting that miR-378* can mediate metabolic in cancer cells [Citation15]. Mice with mutations in miR-378/378* gene are resistant to obesity induced by high fat diet by regulating mitochondrial lipid metabolism [Citation16]. Moreover, miR-378 modulates hepatic insulin signaling by combining with p110α and controlling glucose and lipid homeostasis [Citation14]. Increasing evidence shows that miR-378 plays an important role in the regulation of lipid metabolism and the occurrence and development of liver diseases [Citation17]. Combined with high-throughput sequencing, we previously observed that miR-378b also modulates insulin signaling by specifically binding the insulin receptor and p110α in animal and cellular models of ALD [Citation18]. Along with previous research, we predicted the target of miR-378b via starBase and found that CAMKK2 messenger RNA (mRNA) contains a miR-378b-binding site in its 3ʹ-untranslated region (UTR) which is the upstream of AMPK, suggesting that miR-378b may play an important role in regulating AMPK signal conduction.
Therefore, we hypothesized that miR-378b regulates the pathogenesis of ALD by mediating lipid metabolism via the CaMKK2-AMPK pathway. This study aimed to assess miR-378b expression in the models of ALD in vivo and in vitro, and to further clarify the biological function of miR-378b in EtOH-induced hepatic lipid accumulation, providing direction and target for the mechanism research and drug therapy of ALD.
2. Materials and methods
2.1. Animals and protocols
Male C57BL/6 mice (weighing 20–25 g) were purchased from Hunan Sja Laboratory Animal Co. Ltd (Hunan, China). The animal use program was approved by the Animal Care and Use Committee of Guilin Medical University, and all procedures were in line with the Guidelines for the Care and Use of Experimental Animals issued by the National Institutes of Health. The mice mentioned above were randomly divided into a control group (CD-fed with a control liquid diet) and a model group (fed with a liquid ethanol diet) with 10 mice in each group. The model of alcoholic liver disease (NIAAA model) was constructed with reference to previous methods [Citation19]. All feeds used to feed the mice were purchased from Trophic Animal Feed High-tech Co.LTD (Nantong, China). After four weeks of paired-fed, all the mice were euthanized and their livers and bloods were collected for follow-up tests.
2.2. Adeno-associated virus administration
The complete sequence of mouse miR-378b was amplified from mouse genome DNA by PCR and cloned into pAAV-MCS with double enzyme digestion. Adeno-associated CCTED virus (AAV, serotype 9, a gift from Gene Chem, Shanghai, China) showed higher infection efficiency and induced persistent expression, and was used to package recombinant AAV [Citation20]. The packaged adeno-associated virus was named AAV-miR-378b. The empty (untransformed) adeno-associated virus vector named AAV-NC was used as control. Before the construction of ALD model, the adeno-associated virus vector was injected into the tail vein with 1 ml sterile syringe according to groups, and the dose was 1 × 1011 vg per animal.
2.3. Liver histology
The histopathological analysis was roughly as described previously [Citation21]. Briefly, fixation with formalin buffered with 10% phosphoric acid for 24 h was used to immobilized liver tissue, then embedded with paraffin blocks and stained with hematoxylin and eosin (H&E). Pathological changes were evaluated and micro-graphs were obtained from light microscopes (Olympus BX41, Olympus, Tokyo, Japan).
2.4. Biochemical analysis
Hepatic lipids were extracted as previously reported [Citation22]. Total TG and total cholesterol (TC) were measured using with commercially available diagnostic kits (Unitech Medical Electronics Co., Ltd., Guilin, China).
2.5. Cell culture
L-02 human hepatocytes (No. GDC079) were provided from the Wuhan University (Wuhan, China). The specific methods used for cell culture refer to previous protocols [Citation23]. Moreover, preceding research showed that 200 mM EtOH successfully caused to happen ALD in L-02 cells [Citation18], so our following experiments were performed with an EtOH concentration of 200 mM.
2.6. Plasmid administration
An miR-378b mimic and inhibitor (including their respective negative controls-NCs) were purchased from GeneChem (Shanghai, China). The plasmid was transferred into cells by electroporation (Bio-Rad Laboratories, Inc., Hercules, CA, USA) [Citation18].
2.7. Cellular lipid determination
L-02 cells cultured in a 55 square centimeters petri dish under the nutrient conditions described above were divided into a control group, model group (200 mm EtOH treatment), miR-378b mimic, inhibitor and their corresponding NC group. Except for cells in the control group, others were cultured in a fresh medium containing 200 mM EtOH for 48 h [Citation18]. After cells cultured in groups, intracellular lipids were extracted according to the previously described method [Citation24]. The contents of TG and TC in cells were determined by the kit (Jiancheng Biological Engineering Research Institute Co., Ltd.Nanjing China).
2.8. Western blot analysis and antibodies
Total protein of L-02 cells or liver tissues were extracted using RIPA reagent (Beyotime Bio Co., Nanjing, China). The protein concentration was assessed using the Nanodrop 2000 spectrophotometer (Thermo Scientific, USA). According to the manufacturer’s instructions [Citation18], the same amount of proteins (30 µg) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then attached onto PVDF membranes (Millipore Corp,Billerica, MA, USA). In addition, the rotational conditions are slightly adjusted according to the molecular weight. After transferred, the membrane was closed with 5% skim milk or BSA for 1 h. And then, the membranes were incubated with specific primary antibodies at 4°C overnight or at least 8 h, followed by incubation with anti-rabbit or anti-mouse secondary antibodies conjugated to peroxidase, and the protein bands were then visualized using an enhanced chemiluminescence system (ECL) reagent (E002-100, 7Sea Biotech Co., Shanghai, China). Antibodies against CaMKK2 (DF4793), Carnitine Palmitoyl Transferase 1 (CPT1-DF12004), Peroxisome Proliferator Activated Receptor (PPAR -AF5301), FASN (DF6106), β-actin (AF7018) were obtained from Affinity Biosciences (OH, USA). Antibodies against ACC (C83B10), phosphorylated ACC (GR103168-11), AMPK (GR284787-6), phosphorylated AMPK (GR208507-23) and SREBP1c (GR3202903-3) were obtained from Abcam Technology (Cambridge, UK).
2.9. RNA extraction and real-time PCR
TRIzol reagent (Gibco BRL) was used for extracting total RNA from L-02 cells or the liver. According to the manufacturer’s protocol [Citation25], one microgram of total RNA from each set of samples was used for reverse transcription with a FastQuant RT Kit (B639252, Sangon Biotech Inc., ShangHai, China). Target genes were amplified on the MJ PTC-200 PCR system (Bio-Rad, Hercules, CA, USA) in accordance with the primer sequences listed in . The expression of target genes after amplification was detected by 1% agarose gel electrophoresis and β-actin was used as the reference. MicroRNA was detected by quantitative real-time (qRT)-PCR using a miR-378b specific primer (GeneCopoeia™, Guangzhou, China), and U6 was the reference gene. The dissolution curve, single dominant peak and the stability of the obtained data were comprehensively analyzed to verify the reliability of the strength results.
Table 1. Primer sequences for the associated genes which are tested
2.10. Luciferase reporter assays
A luciferase assay was carried out as described before with some modifications [Citation18]. To clarify whether miR-378 directly combines with CAMKK2, we packaged a plasmid containing luciferase with the 3ʹ-UTR sequence of CaMKK2 mRNA. The targeting relationship between miR-378b and CaMKK2 was determined by cotransfection of pGL3 and PRL-Tk vectors. The relative luciferase activity were determined using The Dual-Luciferase Reporter Assay System (E2920, Promega, Madison, WI, USA).
2.11. Statistical analyses
All data presented are representative of at least 3 repeated experiments and expressed as the mean ± SD. The p < 0.05 was the level of significance. Differences between means were evaluated by analysis of variance (ANOVA) and Student’s t-test. Data analyses and figure preparation were carried out with GraphPad Prism 6.0.
3. Results
In this study, male C57BL/6 mice and L-02 human hepatocytes were chosen and randomly divided into two groups: normal control group and ethanol group. The expression levels of miR-378b in liver tissues and L-02 cells were tested to confirm whether ethanol induced abnormal expression of miR-378b. Subsequently, luciferase reporter assays were analyzed to verify the targeting relationship between miR-378b and CaMKK2. Finally, we explored the effect of miR-378b on ethanol-induced hepatic steatosis by constructing miR-378b adeno-associated virus delivered to ethanol-fed mice or transfecting miR-378b plasmid into ethanol-induced L-02 cells. The expression levels of miR-378b, CaMKK2-AMPK signal, downstream factors associated with lipid synthesis (FASN and SREBP1c) and factors that promote lipid oxidation (PPAR and CPT1) in liver tissues and L-02 cell were tested. We discovered a critical role of miR-378b in the development of ALD.
3.1. miR-378b was over-expressed in cells and animals of ALD models
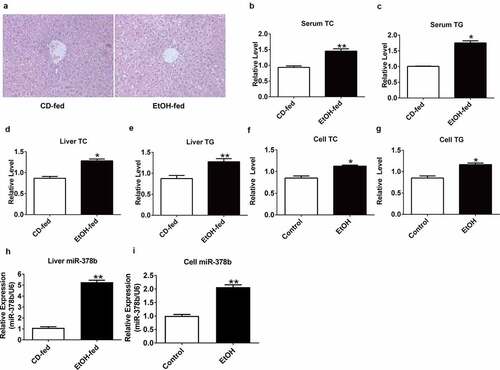
H&E staining was carried out to evaluate the degree of liver injury in mice induced with ethanol. Compared with the livers of mice fed with a control diet, histopathological images showed a significant increase in fat vacuoles in the liver tissues of EtOH-fed mice, accompanied by hepatocyte cord disorder (). To clarify the relationship between ethanol and lipid levels, TG/TC concentrations were determined in the collected mouse liver and serum, the data showed that the serum and liver TC and TG levels of mice fed with ethanol were significantly increased by 41.8% and 61.3% respectively compared with mice fed with control diet (). L-02 cells were treated as described above [Citation18], and the cell contents were obtained for assessment of TC and TG levels, which showed significant differences in the TC and TG content between the groups () by 19.2% and 33.7%, respectively. Next, we examined the miR-378b expression in the cells and animals used to imitate ALD model, and found that the expression of miR-378b was up-regulated compared to that in the control group () by 420% and 87.5%.
Figure 1. miR-378b is up-regulated in EtOH-fed mice and EtOH-induced L-02 cells. (a) Representative images of H&E staining of liver sections from control diet (CD)-fed mice (left) or EtOH-fed mice (right). (b) Serum TC levels. (c) Serum TG levels. (d) Liver TC levels. (e) Liver TG levels. (f) Cell TC levels. (g) Cell TG levels. (h) The expression level of miR-378b in liver tissues. (i) The expression level of miR-378b in L-02 cells. All data are expressed as the mean ± SD of at least three separate experiments (n = 3). *p < 0.05 vs. control; **p < 0.01 vs. control
3.2. miR-378b directly targets CaMKK2
The obvious change in miR-378b expression in the ALD process inspired us to explore the downstream regulators of miR-378b. Through the bio-informatics tools miRBase and TargetScan, we identified CaMKK2 as a potential target of miR-378b with a putative miR-378b-binding site within the 3ʹ-UTR of CaMKK2 ().
Figure 2. miR-378b directly targets CaMKK2. (a) Predicted duplex formation between miR-378b and human wild-type/mutant CaMKK2-3ʹ-UTR. (b) Luciferase reporter assay for the interaction between wild-type 3ʹ-UTR of CaMKK2, as well as mutant 3ʹ-UTR of CaMKK2 in the 293 T cells. (c-d) mRNA expression levels of CaMKK2 in L-02 cells. (e-f) Western blot analysis for protein expression of CaMKK2 in L-02 cells. All data were expressed as the mean ± SD of at least three separate experiments.*p < 0.05,**p < 0.01 vs. control
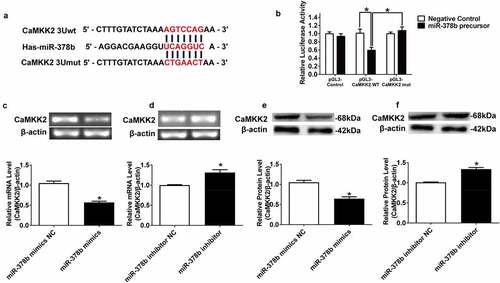
Figure 3. miR-378b over-expression disturbs lipid metabolism in L-02 cells. (a) The TC level in L-02 cells. (b) The TG level in L-02 cells. (c) The expression level of miR-378b in L-02 cells. (d) mRNA expression levels of CaMKK2 in L-02 cells. (e) Western blot analysis for protein expression of CaMKK2 and p-AMPK/AMPK. (f) mRNA expression levels and protein expression levels for PPARα and CPT1. (g) mRNA expression levels and protein expression levels for FASN and SREBP1c. (h) Western blot analysis for protein expression of p-ACC/ACC. All data are expressed as the mean ± SD of at least three separate experiments. *p < 0.05, **p < 0.01 vs. control. #p < 0.05, ##p < 0.01 vs. miR-378b-mimics NC
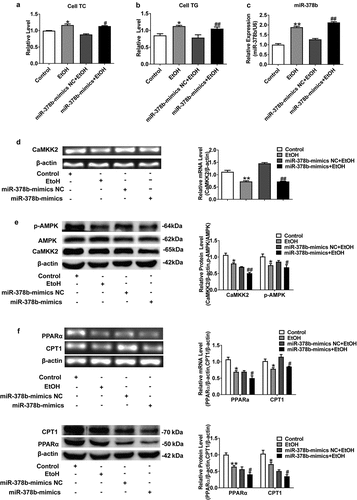
Figure 3. Continued
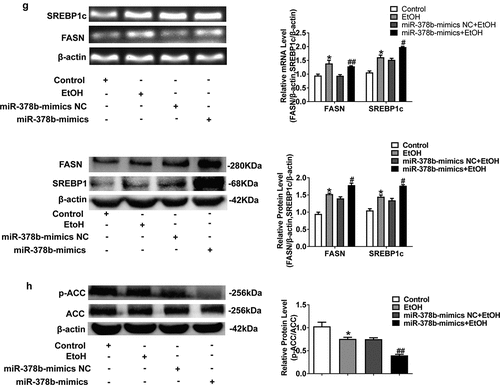
Figure 4. Suppression of miR-378b improved lipid metabolism in L-02 cells. (a) The TC levels in L-02 cells. (b) The TG levels in L-02 cells. (c) The expression level of miR-378b in L-02 cells. (d) The mRNA expression level of CaMKK2 in L-02 cells. (e) Western blot analysis of the protein expression of CaMKK2 and p-AMPK/AMPK protein ratio. (f) The mRNA and protein expression levels of PPARα and CPT1. (g) The mRNA and protein expression levels of FASN and SREBP1c. (h) Western blot analysis of the p-ACC/ACC protein ratio. All data are expressed as the mean ± SD of at least three separate experiments. *p < 0.05, **p < 0.01 vs. control. #p < 0.05, ##p < 0.01 vs. miR-378b-inhibitor NC
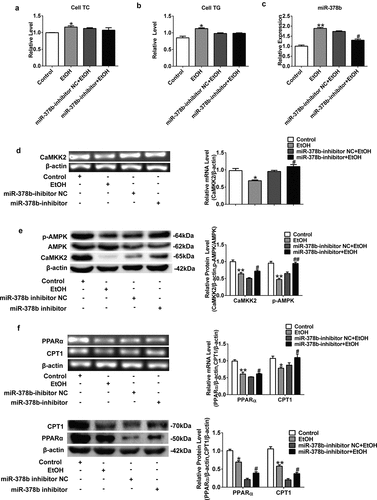
Figure 4. Continued
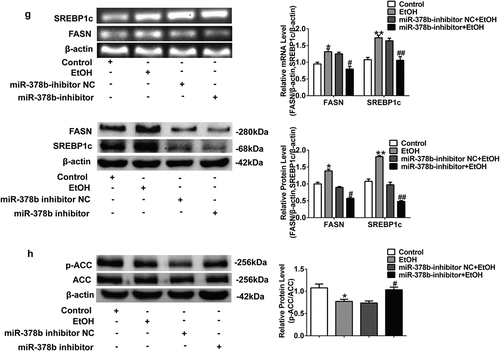
Figure 5. miR-378b over-expression aggravates lipid accumulation in vivo. (a) Representative images of H&E staining of liver sections. (b) Serum TC levels. (c) Serum TG levels. (d) Liver TC levels. (e) Liver TG levels. (f) The expression of miR-378b in liver tissue. (g) mRNA expression levels of CaMKK2 in liver tissue. (h) Western blot analysis for protein expression of CaMKK2 and p-AMPK/AMPK in liver tissue. (i) mRNA expression levels and protein expression levels for PPARα and CPT1 in liver tissue. (j) mRNA expression levels and protein expression levels for FASN and SREBP1c in liver tissue. (k) Western blot analysis for protein expression of p-ACC/ACC in liver tissue. All data were expressed as the mean ± SD of at least three separate experiments (n = 3). *p < 0.05, **p < 0.01 compared with the CD-fed group. #p < 0.05, ##p < 0.01 compared with the AAV-miR-378b-up NC group
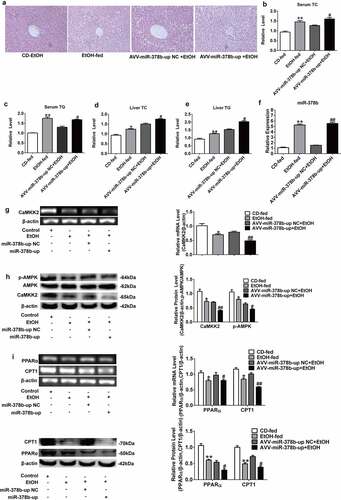
Figure 5. Continued
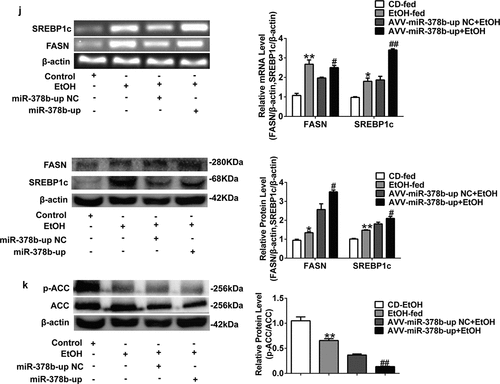
Figure 6. miR-378b inhibition amoliorates lipid lipid dysfunction in vivo. (a) Representative images of H&E staining of liver sections. (b) Serum TC levels. (c) Serum TG levels. (d) Liver TC levels. (e) Liver TG levels. (f) The expression of miR-378b in liver tissue. (g) mRNA expression levels of CaMKK2 in liver tissue. (h) Western blot analysis for protein expression of CaMKK2 and p-AMPK/AMPK in liver tissue. (i) mRNA expression levels and protein expression levels for PPARα and CPT1 in liver tissue. (j) mRNA expression levels and protein expression levels for FASN and SREBP1c in liver tissue. (k) Western blot analysis for protein expression of p-ACC/ACC in liver tissue. All data were expressed as the mean ± SD of at least three separate experiments (n = 3). *p < 0.05, **p < 0.01 compared with the CD-fed group. #p < 0.05, ##p < 0.01 compared with the AAV-miR-378b-down NC group
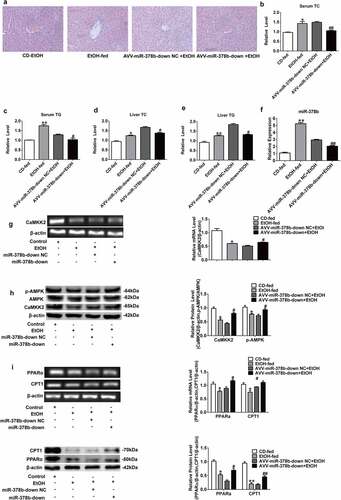
Figure 6. Continued
To determine whether miR-378b directly targets CaMKK2, we performed a luciferase reporting assay. The 3 ‘-UTR of CaMKK2 was cotransfected into 293 T cells with the miR-378b plasmid, and the final results illustrated that miR-378b mimics decreased the luciferase activity in 293 T cells transfected with the reporter involving the human CaMKK2 3′-UTR by 42.5% while there was no marked change on luciferase activity in cells transfected with mutanted CaMKK2 3′-UTR. (). To further clarify whether miR-378b mediates the expression of CaMKK2, miR-378b mimics and inhibitors were transferred into L-02 cells by electroporation transfection, and then the mRNA and protein levels of CaMKK2 were detected. The results displayed that CaMKK2 mRNA level in the L-02 cells was reduced by 44.7% after transfection with miR-378b mimics () and up-regulated by 39.2% () when the cells were transfected with miR-378b inhibitor. The imaging data showed the CaMKK2 protein levels were decreased by 35.6% when the L-02 cells were transfected with miR-378b mimics (), and increased by by 40.4% () when transfected with miR-378b inhibitor. On the basis of these experimental results, we hypothesized that miR-378b may bind CaMKK2 mRNA to regulate translation and transcription of the CaMKK2 protein.
3.3. Over-expression of miR-378b disrupted lipid metabolism in L-02 cells
To study the influence of miR-378b on lipid metabolism, miR-378b mimics were transfected into L-02 cells induced with 200 mM concentration of ethanol. The TC and TG levels in L-02 cells incubated in ethanol and the miR-378b mimics group based on incubated with ethanol were elevated sharply, with the TC level increased by 33.7% and 19.2%, respectively, and the TG level obviously increased by 46.7% and 35.2%, when compared with the corresponding values in the control group (). Moreover, qRT-PCR results showed that miR-378b content was approximately 68% higher in comparison to that in the NC group after transfection (). Furthermore, we analyzed the expression of CaMKK2 and members of the AMPK signaling pathway and discovered that the CaMKK2 mRNA levels () and CaMKK2 protein, p-AMPK/AMPK protein ratio were significantly decreased by 51%, 28.9% and 16.8%, after transfection (). Except for what has been mentioned above, the effects of miR-378b on lipid metabolism in hepatocytes were examined. The mRNA and protein expression levels of PPAR and CPT1 were sharply decreased by approximately 30% upon transfection with miR-378b mimics (). In addition, the mRNA and protein expression levels of FASN and SREBP1c were increased by nearly 33% in the miR-378b over-expressing group compared with the control group (). Moreover, p-ACC/ACC protein ratio was obviously decreased by 45.3% compared to that in the control group (). In summary, these findings suggest that miR-378b over-expression aggravated lipid synthesis and decreased lipid oxidation in L-02 cells incubated with ethanol.
3.4. Suppression of miR-378b alleviated lipid accumulation in vitro
Based on the result described above that lipid disorder was exacerbated when miR-378b is raised in vitro, miR-378b inhibitors were transfected into L-02 cells induced by 200 mM concentration of ethanol by electrical perforation. expressed that the TC and TG levels in the cells did not change significantly after transfection. In addition, compared with that in the control group, the level of miR-378b was significantly reduced by 36.4% after transfection with miR-378b inhibitors (). Furthermore, we analyzed the expression of CaMKK2 and members of the AMPK signaling pathway and found that the mRNA level of CaMKK2 was slightly increased (), while the protein level of CaMKK2 and p-AMPK/AMPK ratio were significantly increased by 40.3% and 46.3%, respectively, in L-02 cells transfected with miR-378b inhibitor compared with cells in the NC group (). Furthermore, the effects of miR-378b on lipid metabolism in hepatocytes were tested. The protein and mRNA expression levels of PPAR and CPT1 were significantly increased by more than 20%, with the protein level increased by more than 50% when cells were transfected with miR-378b inhibitor and incubated in the presence of EtOH (). In addition, the protein and mRNA expression levels of FASN and SREBP1c were decreased by approximately 30% in the miR-378b inhibitor group compared with the control group (). Additionally, the p-ACC/ACC protein ratio was dramatically increased by 30.1% (). In summary, these results indicate that down-regulation of miR-378b reduced lipid synthesis and ameliorated lipid oxidation in L-02 cells incubated with ethanol.
3.5. miR-378b over-expression aggravated lipid accumulation in vivo
To study the influence of miR-378b on lipid metabolism in vivo, we injected the mice used to simulate ALD model with adeno-associated virus particles including AAV-miR-378b-up or corresponding NC. Through qPCR and liver tissue H&E staining, it could be confirmed that the over-expressing miR-378b can induce adipogenesis in liver tissue (). The indicators related lipid such as TC and TG levels in both the serum and liver were increased by nearly 25% in the mice fed with alcohol diet compared to control group (). Moreover, miR-378b expression in the liver of AAV-miR-378b-up-treated mice was approximately twofold higher than that in liver of the NC mice (). In addition, we assessed the levels of proteins involved in lipid metabolism signaling. MiR-378b over-expression reduced CaMKK2 mRNA levels (), CaMKK2 protein expression and the p-AMPK/AMPK protein ratio by at least 29.7% in EtOH-fed mice (). In addition, the mRNA and protein levels of PPAR and CPT1, which are downstream molecules of AMPK, were significantly reduced by at least 20.3% (). Moreover, the mRNA and protein expression of FASN and SREBP1c was also significantly increased by more than 26.2% in mice feed injected with AAV-378b-up compared to those injected with the control (). Beyond that, the p-ACC/ACC protein ratio was decreased by 66.3% upon AAV-miR-378b-up injection of the EtOH-fed mice (). In brief, we concluded that miR-378b over-expression dramatically aggravates lipid accumulation induced by EtOH in vivo.
3.6. The loss of miR-378b alleviated lipid metabolism in mice
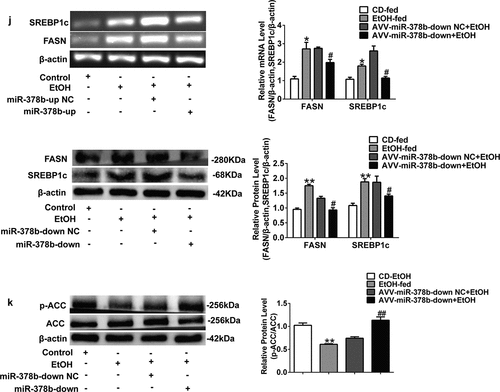
Inhibition of miR-378b reduced lipid accumulation in hepatocytes, then similar results were shown in the mice fed with alcohol. Adeno-associated virus named AAV-miR-378b-down or corresponding control group (NC) was injected into the mice. H&E staining revealed that alcohol induced lipid accumulation in the liver of mice in comparison with the NC, and the degree of hepatic steatosis was significantly improved in EtOH-fed mice infected with AAV-miR-378b-down in comparison with AAV-miR-378b-down-NC mice (). In addition, AAV-miR-378b-down infection led to a obvious reduction in both the hepatic and serum TC and TG content in EtOH-fed mice by at least 20% (). Furthermore, miR-378b expression was decreased by 39.8% in the mice infected AAV-miR-378b-down compared with its′ homologous NC group (). To address the mechanisms underlying miR-378b-mediated regulation of EtOH-induced lipid accumulation, we determined the changes of related lipid metabolism signaling pathways when miR-378b was down-regulated. As expected, inhibition of miR-378b significantly increased CaMKK2 mRNA levels by 28% (), CaMKK2 protein level by 81.8 and p-AMPK/AMPK protein ratio by 27.4%, respectively, in EtOH-fed mice (). Moreover, we assessed PPARα and CPT1 mRNA and protein expression levels in mice, which increased after the injection of AAV-miR-378b-down, with the protein expression level increased nearly one-fold but the mRNA expression increased by approximately 20% (). Moreover, the mRNA and protein expression of FASN and SREBP1c was decreased by at least 26% in EtOH-fed mice after the injection of AAV-miR-378b-down compared to the control (). On the other hand, the p-ACC/ACC protein ratio was increased by 57.5% (). Collectively, these findings indicate that inhibition of miR-378b improved lipid metabolism dysfunction in the livers of EtOH-fed mice.
4. Discussion
Lipid metabolism dysfunction is involved in many metabolic syndromes and induces the over-accumulation of TG when patients present the pathological condition of hepatic steatosis [Citation26]. Furthermore, excessive hepatic fat accumulation can induce metabolic changes, making the liver more sensitive to further injury [Citation27]. Hence, investigating the mechanistic processes governing lipid metabolism might assist us in understanding the pathogenesis of ALD. In this study, we found that miR-378b expression was increased in EtOH-fed mouse livers and EtOH-induced L-02 cells compared to the corresponding control, accompanied by increased lipid accumulation. Furthermore, we focused on whether miR-378b regulates the progression of ALD, and the subsequent investigation showed that miR-378b regulates lipid metabolism by directly targeting CaMKK2 through inhibition of the AMPK cascade. In addition to this, over-expression of miR-378b aggravated lipid accumulation by up-regulating lipogenesis genes and down-regulating fatty acid oxidation genes, while the loss of miR-378b improved lipid metabolism dysfunction in EtOH-fed mouse livers and EtOH-induced L-02 cells. Taken together, both our in vivo and in vitro findings demonstrate that miR-378b plays a role in EtOH-induced hepatic lipid accumulation.
MiRNAs have been shown to regulate lipid metabolism directly or indirectly through epigenetic mechanisms [Citation17]. Previous research showed that miR-378b was markedly decreased in plasma from coronary heart disease (CHD) patients, the livers from diet-induced obese SD rats under hypoxic training and neurofibromatosis 1 (NF1) patient brain specimens [Citation28,Citation29], and miR-378b expression was also significantly increased in luteinizing hormone-treated mouse TM3 Leydig cells [Citation30]. In this paper, we took the lead in finding that miR-378b was up-regulated by EtOH, and miR-378b was involved in hepatic lipid dysfunction induced by EtOH.
CaMKK2 (a 66–68-kDa kinase) is one of the most versatile CaMKs and participates in mediating many important physiological and pathophysiological processes and conditions, including energy balance, adiposity, glucose homeostasis, hematopoiesis, inflammation, and cancer [Citation8]. A previous study showed that deletion of CAMKK2 in vivo or in human prostate cancer cells reduced the expression of two key lipogenic enzymes, ACC and FASN, and finally led to inhibition of de novo lipogenesis [Citation31]. Primary hepatocytes from CaMKK2-knockout (KO) mice showed suppressed gluconeogenesis and increased de novo lipogenesis and fatty acid oxidation, and an inhibitor of CaMKK2 improved hepatic steatosis and ameliorated the symptoms of nonalcoholic fatty liver disease (NAFLD) [Citation10,Citation13]. However, our understanding of the physiological function of CaMKK2 in EtOH-induced lipid accumulation is limited. In this study, CaMKK2 was identified as a miR-378b target gene by using TargetScan. Luciferase constructs harboring the 3ʹ-UTRs of CaMKK2 showed significantly decreased activity in 293 T cells transfected with miR-378b mimics. In order to verify the regulatory relationship between miR-378b and CaMKK2, we up-regulated or down-regulated the expression of miR-378b in L-02 cells and found that miR-378b over-expression led to a decrease in CaMKK2, and down-regulation of miR-378b increased CaMKK2 expression. Thus, CaMKK2 is clearly the direct target of miR-378b. We also found that the up-regulation of miR-378b in EtOH-induced L-02 cells sharply inhibited CaMKK2 protein level and exacerbated lipid accumulation, possibly associated with increased lipogenic gene expression or decreased lipid oxidative gene expression. However, inhibition of miR-378b enhanced the CaMKK2 protein level and improved lipid metabolism disorder. These findings further support the function of miR-378b to hepatic lipid metabolism through CaMKK2. However, the function of CaMKK2 in lipid metabolism appears to differ in different diseases (mentioned above), and how it definitively functions in different diseases and whether its function is time-dependent need to be explored.
Human miR-378b significantly increases in level during keratinocyte differentiation and inhibits the proliferation, migration and differentiation of keratinocytes [Citation32]. In addition, miR-378b was proved to be able to activate the cGAS/STING signaling pathway and regulate dendritic cell (DC) function by binding TANK-binding kinase binding protein 1 (TBKBP1) [Citation33]. However, the function of miR-378b in the pathogenesis of hepatic lipid metabolism has not been elucidated. To clarify this, we over-expressed or silenced miR-378b in L-02 cells. Our data showed that the up-regulation of miR-378b in L-02 cells dramatically increased the TC and TG contents, but the inhibition of miR-378b did not significantly change the TC or TG levels. The multi-program nature of TG synthesis may be one of the reasons to explain this phenomenon, because it takes some time to complete the progress which start form regulation of miRNA level to obvious change of lipid level. In addition, ethanol and miR-378b over-expression had the similar superposition effect, while miR-378b inhibition was corresponding to ethanol. Therefore, it may take longer time to reverse ethanol-induced lipid accumulation when L-02 cells were transfected with miR-378b inhibitor than miR-378b mimics, which has been reflected in animal experiments with four weeks. On the basis of this, we examined the CaMKK2 and the AMPK cascade, which are related to lipid metabolism. Our data showed that the miR-378b over-expressed in L-02 cells greatly inhibited the protein of CaMKK2, p-AMPK, p-ACC and enzymes related to lipid oxidation (PPARα and CPT1) and even increased the expression of de novo lipogenesis-related proteins (FASN and SREBP1c). However, the inhibition of miR-378b increased the expression level of CaMKK2 and improved lipid metabolism dysfunction. These findings indicate that miR-378b regulated EtOH-induced hepatic steatosis by increasing lipogenesis and decreasing fatty acid oxidation in L-O2 cells. Briefly, our in vitro research illustrated that miR-378b is involved in lipid metabolism by targeting CaMKK2 to regulate the AMPK cascade. However, we noted no obvious change in the TC or TG content in L-02 cells after transfection with miR-378b inhibitor. While the reason for this remains unknown, many factors could be involved, such as the method of detection and number of cells.
Then, we established the NIAAA mouse model through feeding a Lieber-DeCarli LD supplemented with 5% EtOH. We confirmed the over-expression of miR-378b in EtOH-fed mouse livers compared to those of the NC group. Moreover, we also observed increases in TG and TC levels in the liver and serum of EtOH-fed mice compared to mice in the NC group, indicating lipid dysfunction in the liver. Then, we carried out functional experiments using miR-378b. After tail vein injection of AAV-miR-378b or AAV-NC, the expression of miR-378b was increased in the livers of AAV-miR-378b mice relative to those of mice in the AAV-NC group. Remarkably, the over-expression of hepatic miR-378b increased TC and TG levels in both the liver and serum and aggravated lipid accumulation in accordance with inhibition of CaMKK2 and the AMPK cascades. The loss of miR-378b notably decreased TC and TG levels and ameliorated lipid accumulation induced by EtOH by up-regulating CaMKK2. In summary, our results indicate that the up-regulation of miR-378b accelerated hepatic lipid accumulation in EtOH-fed mice by increasing lipid synthesis and decreasing lipid oxidation and that inhibition of miR-378b improved lipid metabolism disorder induced by EtOH in vivo. Although we determined the function of miR-378b in ALD, whether EtOH interacts with miR-378b directly or indirectly remains unknown. In summary, miR-378b is a crucial regulator of ALD in vitro through its mediation of lipid metabolism.
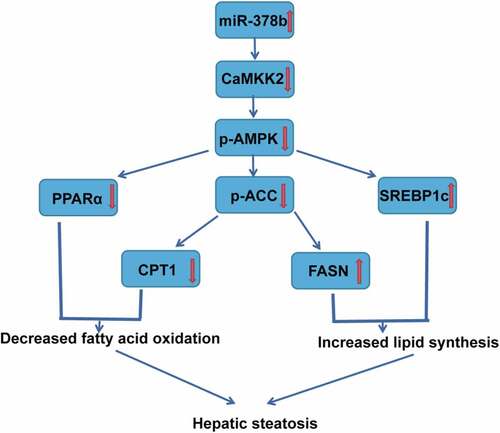
We can thus conclude () that excessive EtOH consumption can lead to high miR-378b expression. The CaMKK2 mRNA 3-UTR contains a miR-378b-binding site, so miR-378b does influence the stability of CaMKK2 mRNA and the translation of CaMKK2 mRNA, down-regulating the CaMKK2 protein in the liver and decreasing AMPK phosphorylation, which disturbs the lipid metabolism cascade in the liver. Because of inhibition of the AMPK pathway, lipid synthesis in the liver is increased, and lipid oxidation is decreased, eventually causing fatty liver. In brief, up-regulation of miR-378b expression in the liver is deeply involved in ALD formation. Therefore, miR-378b is a potential future target for ALD treatment. Next, as described above, we will continue to explore the mechanism by which EtOH interacts with miR-378b and investigate the degree to which miR-378b regulates lipid accumulation by targeting CaMKK2 in an ALD model. Moreover, if possible, it would be beneficial for us to detect the expression of miR-378b, CaMKK2 and members of the AMPK cascade in clinical specimens.
Figure 7. Summary of the mechanism by which miR-378b targets CaMKK2 to regulate hepatic steatosis through the AMPK cascade. First, the increased expression of miR-378b induced by EtOH decreases the expression of CaMKK2, after which the AMPK cascade is inhibited, eventually aggravating steatosis
5. Conclusions
Our results suggest that alcohol-abuse disturbs the lipid metabolism in the liver by miR-378b up-regulation. The over-expression of miR-378b exacerbates the lipid accumulation in ethanol-induced hepatocytes and liver, while miR-378b silencing inhibits ethanol-induced hepatic steatosis to some extent. Establishing the role of miR-378b in EtOH-induced hepatic steatosis could possibly pave the way for new direction and target for the mechanism research and drug therapy of ALD.
Author contributions
Ying-zhao Wang, Jun Lu: Data curation,Writing- Original draft preparation.
Yuan-yuan Li, Yu-juan Zhong, Cheng-fang Yang, Yan Zhang: Conceptualization, Methodology. Li-hua Huang, Su-mei Huang,: Supervision. Qi-ran L, Dan Wu, Meng-wei Song, Lin Shi: Software, Validation. Yong-wen Li, Li Li: Project administration, Funding acquisition.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NO. 81760669; 82060673; 81860660), the Guangxi Natural Science Foundation Project of Guangxi Province, China (No. 2018GXNSFDA281012), Innovation Projiect of Guangxi Graduate Education (YCSW2021257).
Disclosure statement
The authors report no conflict of interest.
Additional information
Funding
References
- Fuster D, Samet JH. Alcohol use in patients with chronic liver disease. N Engl J Med. 2018;379(13):1251–1261.
- Axley PD, Richardson CT, Singal AK. Epidemiology of alcohol consumption and societal burden of alcoholism and alcoholic liver disease. Clin Liver Dis. 2019;23(1):39–50.
- Mitra S, De A, Chowdhury A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl Gastroenterol Hepatol. 2020;5:16.
- Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141(5):1572–1585.
- Baghy K, Iozzo RV, Kovalszky I. Decorin-TGFβ axis in hepatic fibrosis and cirrhosis. J Histochem Cytochem. 2012;60(4):262–268.
- Jiang Z, Zhou J, Zhou D, et al. The adiponectin-SIRT1-AMPK pathway in alcoholic fatty liver disease in the rat. Alcohol Clin Exp Res. 2015;39(3):424–433.
- Singal AK, Bataller R, Ahn J, et al. ACG clinical guideline: alcoholic liver disease. Am J Gastroenterol. 2018;113(2):175–194.
- Racioppi L, Means AR. Calcium/Calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J Biol Chem. 2012;287(38):31658. 10.1074/jbc.R112.356485.
- Chauhan AS, Liu X, Jing J, et al. STIM2 interacts with AMPK and regulates calcium-induced AMPK activation. FASEB J. 2019;33(2):2957–2970.
- Wang Q, Liu S, Zhai A, et al. AMPK-mediated regulation of lipid metabolism by phosphorylation. Biol Pharm Bull. 2018;41(7):985–993. 10.1248/bpb.b17-00724.
- Garcia D, Shaw RJ. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell. 2017;66(6):789–800.
- Li Y, Xu S, Mihaylova MM, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13(4):376–388.
- Jadeja RN, Chu X, Wood C, et al. M3 muscarinic receptor activation reduces hepatocyte lipid accumulation via CaMKKβ/AMPK pathway. Biochem Pharmacol. 2019;169:113613. 10.1016/j.bcp.2019.08.015.
- Liu W, Cao H, Ye C, et al. Hepatic miR-378 targets p110α and controls glucose and lipid homeostasis by modulating hepatic insulin signaling. Nat Commun. 2014;5(1):5684.
- Eichner LJ, Perry MC, Dufour CR, et al. miR-378(*) mediates metabolic shift in breast cancer cells via the PGC-1β/ERRγ transcriptional pathway. Cell Metab. 2010;12(4):352–361.
- Carrer M, Liu N, Grueter CE, et al. Control of mitochondrial metabolism and systemic energy homeostasis by microRNAs 378 and 378*. Proc Natl Acad Sci U S A. 2012;109(38):15330–15335.
- Yang Z, Cappello T, Wang L. Emerging role of microRNAs in lipid metabolism. Acta Pharm Sin B. 2015;5(2):145–150.
- Li YY, Zhong YJ, Cheng Q, et al. miR-378b regulates insulin sensitivity by targeting insulin receptor and p110α in alcohol-induced hepatic steatosis. Front Pharmacol. 2020;11:717.
- Ghosh Dastidar S, Warner JB, Warner DR, et al. Rodent models of alcoholic liver disease: role of binge ethanol administration. Biomolecules. 2018;8(1):3.
- Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet. 2011;12(5):341–355.
- Li C, Li L, Yang CF, et al. Hepatoprotective effects of Methyl ferulic acid on alcohol-induced liver oxidative injury in mice by inhibiting the NOX4/ROS-MAPK pathway. Biochem Biophys Res Commun. 2017;493(1):277–285.
- Cheng Q, Li YW, Yang CF, et al. Methyl ferulic acid attenuates ethanol-induced hepatic steatosis by regulating AMPK and FoxO1 pathways in rats and L-02 cells. Chem Biol Interact. 2018;291:180–189.
- Li L, Zhong Y, Ma Z, et al. Methyl ferulic acid exerts anti-apoptotic effects on L-02 cells via the ROS-mediated signaling pathway. Int J Oncol. 2018;53(1):225–236.
- Toppo E, Darvin SS, Esakkimuthu S, et al. Effect of two andrographolide derivatives on cellular and rodent models of non-alcoholic fatty liver disease. Biomed Pharmacother. 2017;95:402–411.
- Yang C, Li L, Ma Z, et al. Hepatoprotective effect of methyl ferulic acid against carbon tetrachloride-induced acute liver injury in rats. Exp Ther Med. 2018;15(3):2228–2238.
- Wang K. Molecular mechanism of hepatic steatosis: pathophysiological role of autophagy. Expert Rev Mol Med. 2016;18:e14.
- You M, Arteel GE. Effect of ethanol on lipid metabolism. J Hepatol. 2019;70(2):237–248.
- Lu YL, Jing W, Feng LS, et al. Effects of hypoxic exercise training on microRNA expression and lipid metabolism in obese rat livers. J Zhejiang Univ Sci B. 2014;15(9):820–829.
- Su M, Niu Y, Dang Q, et al. Circulating microRNA profiles based on direct S-Poly(T)Plus assay for detection of coronary heart disease. J Cell Mol Med. 2020;24(11):5984–5997.
- Li C, Gao S, Chen S, et al. Differential expression of microRNAs in luteinising hormone-treated mouse TM3 Leydig cells. Andrologia. 2018;50(1):e12824.
- Hawley SA, Pan DA, Mustard KJ, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2(1):9–19.
- Wang XL, Zhang T, Wang J, et al. MiR-378b promotes differentiation of keratinocytes through NKX3.1. PloS One. 2015;10(8):e0136049.
- Shah AU, Cao Y, Siddique N, et al. miR29a and miR378b influence CpG-stimulated dendritic cells and regulate cGAS/STING pathway. Vaccines (Basel). 2019;7(4):197.