
ABSTRACT
Acute kidney injury (AKI), a common complication of sepsis, is characterized by a rapid loss of renal excretory function. A variety of etiologies and pathophysiological processes may contribute to AKI. Previously, mitogen-activated protein kinase 1 (MAPK1) was reported to regulate cellular processes in various sepsis-associated diseases. The current study aimed to further explore the biological function and regulatory mechanism of MAPK1 in sepsis-induced AKI. In our study, MAPK1 exhibited high expression in the serum of AKI patients. Functionally, knockdown of MAPK1 suppressed inflammatory response, cell apoptosis in response of lipopolysaccharide (LPS) induction in HK-2 cells. Moreover, MAPK1 deficiency alleviated renal inflammation, renal dysfunction, and renal injury in vivo. Mechanistically, MAPK1 could activate the downstream p38/NF-κB pathway. Moreover, long noncoding RNA potassium voltage-gated channel subfamily Q member 1 opposite strand/antisense transcript 1 (KCNQ1OT1) was identified to serve as a competing endogenous RNA for miR-212-3p to regulate MAPK1. Finally, rescue assays indicated that the inhibitory effect of KCNQ1OT1 knockdown on inflammatory response, cell apoptosis, and p38/NF-κB pathway was reversed by MAPK1 overexpression in HK-2 cells. In conclusion, KCNQ1OT1 aggravates acute kidney injury by activating p38/NF-κB pathway via miR-212-3p/MAPK1 axis in sepsis. Therefore, KCNQ1OT may serve as a potential biomarker for the prognosis and diagnosis of AKI patients.
Introduction
Sepsis is a systemic inflammatory response syndrome stimulated by infection [Citation1,Citation2]. As a serious complication of sepsis, acute kidney injury (AKI) can result in organ dysfunction and cause high morbidity and mortality in most septic shock patients [Citation3]. Clinically, AKI can be grouped into three primary etiologies: prerenal, renal, and postrenal [Citation4,Citation5]. Various causes of AKI include sepsis, nephrotoxic drugs, dehydration, renal surgery, renal ischemia, ischemia-reperfusion renal injury and urinary tract obstruction [Citation4]. Due to the complexity and uniqueness of sepsis, sepsis-induced AKI is distinct from other types of AKI, which is the first cause of AKI in intensive care unit [Citation6]. Potential pathogenic mechanism underlying sepsis-induced AKI includes cellular metabolic reprogramming, a dysregulated inflammatory response, and microcirculatory dysfunction [Citation7]. At the cellular level, the epithelial-mesenchymal transition (EMT) and the endothelial-to-mesenchymal transition (EndMT) play vital roles in the development of kidney fibrosis [Citation8]. Novel signaling molecules that regulate kidney fibrosis include endothelial glucocorticoid receptors, endothelial Sirtuin 3, endothelial fibroblast growth factor receptor 1 in kidney and podocyte-glucocorticoid receptor signaling in protecting glomerula [Citation9–12]. With the emerging of detection techniques for sepsis-associated AKI, such as evaluation of a urine microscopy score and investigation of novel serum biomarkers, people have more understanding of sepsis-induced AKI [Citation13]. Blood purification and pharmacologic therapies have become promising experimental therapies in recent years [Citation14]. Potential drugs against AKI and renal fibrosis are known as DPP-4 inhibitor linagliptin, empagliflozin, Sirtuin 3, JAK/STAT inhibitors, glycolysis inhibitors, angiotensin-converting enzyme inhibitors, angiotensin receptor antagonists, and peptide AcSDKP [Citation15–21]. However, effective therapeutic strategies for prevention and treatment of sepsis-associated AKI are still limited [Citation14,Citation22]. Therefore, providing new insights into molecular regulatory mechanism associated with AKI is critical to the development of potential therapeutic approaches for AKI.
Many molecules have been reported to be associated with renal injury and fibrosis [Citation23]. For example, SIRT3, an important member of the sirtuin family, can alleviate AKI by improving mitochondrial function and regulating fatty acid oxidation [Citation17,Citation24]. Mitogen-activated protein kinase 1 (MAPK1), also known as p38 or ERK, belongs to the MAP kinase family [Citation25,Citation26]. MAP kinases or ERKs were reported to function as an integration point for several biochemical signaling pathways and involve in various cellular processes like apoptosis, inflammation, and transcription regulation [Citation27–30]. The phosphorylation of upstream kinases could activate MAP kinases. Upon activation, MAP kinase translocates to the nucleus to phosphorylate nuclear targets such as p38 or NF-κB [Citation28,Citation31–33]. This regulatory network of MAPK1 has been identified in various diseases [Citation34–36]. Additionally, the upregulation of miRNA-433-5p activates MAPK1 to protect acute spinal cord injury [Citation37]. MiR-342 suppresses MAPK1 expression to relieve lipopolysaccharide-induced acute lung injury [Citation38]. Guanine-nucleotide exchange factor H1 was proposed to upregulate lipopolysaccharide (LPS)-induced inflammatory cytokines in endothelial cells by activating nuclear factor κB [Citation39]. However, the upstream mechanism of MAPK1 in AKI deserves a further exploration.
MicroRNAs (miRNAs) are highly conserved non-coding transcripts, approximately 18–24 nucleotides in length, which bind to the 3′-UTR of mRNAs to affect their expression in various biological processes [Citation40,Citation41]. Emerging evidence suggests that the alterations of miRNAs can cause fibrotic disorder in kidney [Citation42]. miR200 family and miR205 are highly associated with EMT, and suppression of upregulated miR-21 or overexpression of downregulated miR-125b is a promising approach to inhibit the induction of EndMT [Citation42]. miR-29 family and miR-let-7 family are major anti-fibrotic players in kidney fibrosis [Citation43]. Their participation in crosstalk mechanisms is crucial for endothelial cell homeostasis. miR-29 directly targets DDP-4, and DDP-4 inhibition can suppress EndMT-driven TGFβ signaling in streptozotocin-induced renal fibrosis. Thus, DPP-4 inhibition can be one of therapeutic strategies for diabetic nephropathy [Citation43]. miR-29 and miR-let-7 families exhibit crosstalk regulation by stimulating FGFR1 phosphorylation and targeting TGFβR1 [Citation43]. miR-22-3p targets PTEN to inhibit the inflammatory response and HK-2 cell apoptosis [Citation44]. miR-212-3p has also been documented to restrain inflammatory response by targeting high mobility group box 1 (HMGB1) in LPS-induced murine macrophages [Citation45]. Nevertheless, the interaction between MAPK1 and miR-212-3p is unclear.
Long non-coding RNAs (lncRNAs) are a kind of transcripts with over 200 nucleotides without protein-coding ability [Citation46–48]. LncRNAs can serve as a competing endogenous RNA (ceRNA) to indirectly regulate downstream messenger RNAs by interacting with miRNAs, thus influencing disease phenotypes in kidneys [Citation49]. For example, lncRNA TCONS_00016406 interacts with miR-687 to regulate the expression of phosphatase and tensin homolog (PTEN), thereby attenuating LPS-stimulated AKI, as shown by alleviation of inflammation, the inhibition of oxidative stress and the suppression of cell apoptosis [Citation50]. LncRNA SNHG14 promotes the production of inflammatory cytokines in LPS-induced HK-2 cells by interacting with miR-495-3p to regulate downstream homeodomain interacting protein kinase 1 (HIPK1)/NF-κB/p65 signaling pathway [Citation51]. Potassium voltage-gated channel subfamily Q member 1 opposite strand/antisense transcript 1 (KCNQ1OT1) was reported to exert effects on sepsis-associated diseases including myocardial injury, acute respiratory distress syndrome and preeclampsia in response of LPS treatment [Citation52–54]. Yet, neither the role nor the mechanism of KCNQ1OT1 has been explored in AKI.
The study aimed to explore the role of MAPK1 and its upstream signaling in sepsis-induced AKI. We hypothesized that KCNQ1OT1 might act as a competing endogenous RNA (ceRNA) against miR-212-3p to regulate MAPK1 expression, thereby affecting AKI progression. The study might provide novel biomarkers for the diagnosis and treatment of AKI.
Materials and methods
Cell culture
Human renal tubular epithelial cells (HK-2) were purchased from Cell Bank of Type Culture Collection (Chinese Academy of Sciences, Shanghai, China) and were cultured in RPMI-1640 medium (Gibco, New York, CA, USA) containing 100 mg/ml streptomycin, 100 U/ml penicillin and 10% fetal bovine serum (FBS; Gibco) in a humidified atmosphere at 37°C with 5% CO2.
Cell treatment
To establish an in vitro model of sepsis, lipopolysaccharide (LPS; 1 μg/ml; Sigma, St. Louis, MO, USA) was used to induce inflammation for 24 h. Cells were then inoculated into 60 mm culture plate (1 × 106 cells/ml) for 16 h of incubation at 37°C. Cells in the control group were treated with the same dose of normal saline (Gibco).
Cell transfection
Short hairpin RNA (sh-RNA) targeting MAPK1 (sh-MAPK1) or KCNQ1OT1 (sh-KCNQ1OT1) and negative controls (sh-NC), MAPK1 overexpression vector (pcDNA3.1/MAPK1) and the negative control (empty pcDNA3.1 vector), miR-212-3p mimics/inhibitor and the negative control (NC mimics/inhibitor) were obtained from GenePharma (Shanghai, China). Sequences of plasmids used for cell transfection are provided in . HK-2 cells were seeded in 6-well plates and grown for 24 h until the cell density reached 30–50%. Later, Lipofectamine 2000 (Thermo Fisher Scientific, USA) was used to transfect shRNAs (50 nM), mimics/inhibitors (40 nM) and pcDNA3.1 vectors (10 nM) into HK-2 cells according to the manufacturer’s protocols. After 48 hours, the efficiency of cell transfection was verified by RT-qPCR.
Table 1. Sequences of plasmids used for cell transfection
Animal model
Adult male SpragueDawley (SD) rats (n = 32, 220–250 g) were purchased from Vital River Co. Ltd (Beijing, China). After being anesthetized through intraperitoneal injection of 10% chloral hydrat, rat was cut open on the midline of the abdomen and then separated the mesentery and cecum. Afterward, we ligated the ileocecal valve and punctured the end of cecum twice with needle. The cecum was returned into the abdominal cavity after defecating. In the sham group, the skin was sutured after only separating the mesentery and cecum. The levels of blood urea nitrogen (BUN) and serum creatinine (SCR) were determined by available kits (Thermo Fisher Scientific). After rats were sacrificed 2 weeks after the establishment of the in vivo model, blood was collected, and kidneys were isolated for further study. Animal experiments were performed in reference to a previous study [Citation55].
Preparation and injection of recombinant adeno-associated virus (rAAV)
To manipulate the MAPK1 expression in vivo, the rAAV system (serotype 9) were employed [Citation56]. To knockdown MAPK1 expression, the sh-RNA sequence targeting MAPK1 was amplified by PCR, and then ligated into rAAV vectors. Two weeks before animal model establishment, rats were injected with AAV-sh-MAPK1 (1 × 1012 copies) or AAV-sh-NC (1 × 1012 copies) via tail vein.
Hematoxylin-eosin (HE) staining
HE staining was conducted as previously described [Citation57]. In short, paraffin-coated renal tissue sections (5 μm) were dewaxed and hydrated with xylene. Next, tissue sections were stained with hematoxylin and eosin solution. After dehydration and sealing, the pathological morphology of renal tissue was visualized by an optical microscope.
Enzyme-linked immunosorbent assay (ELISA)
As previously reported, 3 mL blood was collected from rats after anesthesia [Citation57]. After blood samples were centrifuged for 10 min at 3500 r/min, concentrations of TNF-α, IL-6 and IL-1β in the serum were examined using ELISA kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s recommendations.
Flow cytometry analysis
Apoptosis of HK-2 cells was measured utilizing Annexin V-FITC apoptosis detection kit (BD Biosciences; San Jose, CA, USA) according to a previous study [Citation58]. After washing, HK-2 cells were stained by binding buffer including Annexin V-FITC and propidine iodide (PI) in the darkness at 4°C. Afterward, cell apoptosis was evaluated by flow cytometry (FACScan; BD Biosciences, USA). The FITC Annexin V+/PI− and FITC Annexin V+/PI+ cell populations were considered as apoptotic cells.
Immunofluorescence assay
To explore the nuclear transport level of NF-κB, immunofluorescence assay was performed [Citation59]. HK-2 cells were immobilized with 4% paraformaldehyde. Next, cells were permeabilized through 0.1% Triton X-100 and 0.1% Sodium Citrate. Then, HK-2 cells were incubated with anti-NF-κB (ab32536, 1:250; Abcam, Cambridge, MA, USA) for 2 h following by suitable secondary antibody. DAPI (Sigma-Aldrich) was used for nucleus staining. Finally, the fluorescence microscopy (Olympus America Inc, Center Valley, PA) was used to observe cells.
Luciferase reporter assay
The full length of KCNQ1OT1 or 3′-UTR of MAPK1 was cloned into the pmirGLO reporters to construct KCNQ1OT1‐wild type (WT) or MAPK1‐WT reporters. The MAPK1‐mutant (Mut) and KCNQ1OT1‐Mut reporters were also established. MiR‐212-3p mimics/inhibitor (or NC mimics/inhibitor) were co-transfected with plasmids mentioned above into HK-2 cells using Lipofectamine 2000 (Thermo Fisher Scientific). After 48 h of transfection, the luciferase activities were examined by Dual-Luciferase Reporter System (Promega, Madison WI, USA). The firefly luciferase activity was normalized to that of Renilla luciferase. The assay was performed according to the previous study [Citation58].
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
The RNA from rat renal tissues or HK-2 cells was extracted by TRIzol regent (Invitrogen, USA) and reverse transcribed into cDNAs using PrimeScript RT reagent kits (Takara Biotechnology Ltd., Dalian, China). The SYBR® Premix Ex TaqTM II reagent kit (RR820A, Takara) under ABI7500 real-time qPCR system (7500, ABI Company, Oyster Bay, NY, USA) was used for performing qPCR. Gene expression was calculated by 2−ΔΔCT method [Citation60]. GAPDH was an internal reference for MAPK1 and lncRNAs, and U6 was a control for miRNAs.
Western blotting
The protein contents were extracted from rat renal tissues or HK-2 cells with RIPA lysate (Beyotime Biotechnology, Shanghai, China). Then, protein samples were separated by 10% SDS-PAGE and moved to polyvinylidene difluoride membranes. After being blocked with skim milk, the membranes were incubated with the primary antibodies at 4°C overnight. Primary antibodies include anti-Bax (ab182733, 1:2000), anti-Bcl-2 (ab182858, 1:2000), anti-cleaved-caspase-3 (ab2302, 1:500), anti-E-cadherin (ab1416, 1:50), anti-collagen I (ab34710, 1:1000), anti-p38MAPK (ab170099, 1:1000), anti-p-p38MAPK (ab178867, 1:1000), anti-NF-κB (ab32536, 1:1000), anti-p-NF-κB (ab76302, 1:1000), and anti-GAPDH (ab9485, 1:2500). Next, secondary antibodies were added to membranes for another 2 h of incubation at room temperature. The bands were assessed through the ECL chemiluminescent detection system (Thermo Fisher Scientific) and quantified by ImageJ software (National Institutes of Health, Bethesda, MA, USA) [Citation61].
Statistical analysis
All data were shown as mean ± standard deviation (SD) and analyzed by SPSS 19.0 (SPSS, Chicago, IL) [Citation62]. The student’s t-test was applied for two-group comparisons, and one-way analysis of variance followed by Tukey’s post hoc analysis was applied for multiple-group comparisons. A value of p < 0.05 was considered as statistically significant.
Results
Sepsis-induced AKI is the major cause of AKI in intensive care unit. The exploration of biomarkers for sepsis-associated AKI and their underlying mechanism are necessary for the enrichment of therapeutic approaches. Here, MAPK1 was found to be upregulated in LPS-treated HK-2 cells. MAPK1 knockdown inhibited inflammatory response in LPS-induced HK-2 cells and suppressed cell apoptosis. Additionally, silencing MAPK1 downregulated protein levels of EMT marker (E-cadherin) and mesenchymal marker (collagen I) in LPS-treated HK-2 cells. Moreover, silencing MAPK1 inactivated MAPK/NF-κB pathway in LPS-induced HK-2 cells and alleviated sepsis-induced kidney injury in vivo. miR-212-3p was verified to be the upstream molecule of MAPK1. KCNQ1OT1 interacts with miR-212-3p to regulate MAPK1 expression. In rescue experiments, overexpressed MAPK1 reversed the suppressive effect of KCNQ1OT1 knockdown on HK-2 cell injury and p38/NF-κB signaling. Overall, KCNQ1OT1 aggravates sepsis-induced AKI by activating p38/NF-κB signaling via miR-212-3p/MAPK1 axis.
Knockdown of MAPK1 suppresses inflammatory response in LPS-induced HK-2 cells
We first focused on the role of MAPK1 in sepsis-induced AKI. According to RT-qPCR, MAPK1 level was increased by LPS treatment in HK-2 cells (). The knockdown efficiency of the plasmid sh-MAPK1 is confirmed in . Next, through ELISA, the increase in TNF-α, IL-6 and IL-1β concentration was discovered in response of LPS treatment, which was counteracted by silencing MAPK1 (). In general, MAPK1 knockdown inhibits inflammatory response in LPS-induced HK-2 cells.
Figure 1. MAPK1 knockdown suppresses inflammatory response in LPS-induced HK-2 cells
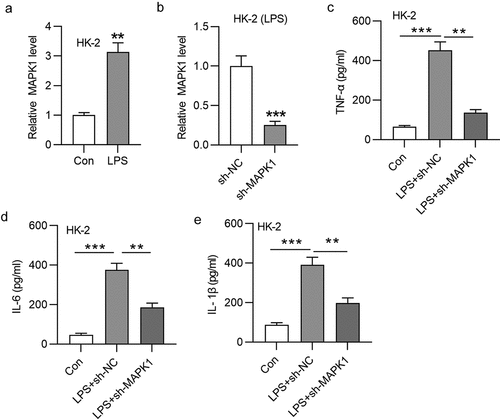
Silencing MAPK1 inhibits HK-2 cell apoptosis and inactivates p38/NF-κB signaling pathway
Subsequent work was done to explore the effect of MAPK1 on cell apoptosis. As displayed in , LPS treatment significantly enhanced the apoptosis of HK-2 cells, but this change could be reversed after silencing MAPK1. Western blotting was performed to examine protein levels of apoptosis markers (Bax, Bcl-2, and cleaved caspase-3), EMT marker (E-cadherin), and fibrosis marker (collagen I) in HK-2 cells with indicated transfection. The results demonstrated that the LPS treatment decreased Bcl-2 protein level but increased Bax and Cleaved caspase-3 protein levels (). Moreover, protein level of E-cadherin was reduced and that of collagen I was increased in HK-2 cells stimulated by LPS (). All these alterations were reversed by MAPK1 knockdown (). MAPK1 is involved in the activation of many pathways, among which p38/NF-κB pathway has been reported to be associated with cell apoptosis and inflammatory response. Western blotting revealed that p-p38MAPK and p-NF-κB protein levels were enhanced in response of LPS treatment and then reduced in LPS-treated HK-2 cells transfected with sh-MAPK1 (). In addition, knockdown of MAPK1 also repressed the nuclear translocation of NF-κB induced by LPS treatment (). To sum up, silencing MAPK1 inhibits HK-2 cell apoptosis and inactivates p38/NF-κB pathway.
Figure 2. Silencing MAPK1 inhibits HK-2 cell apoptosis and inactivates p38/NF-κB pathway
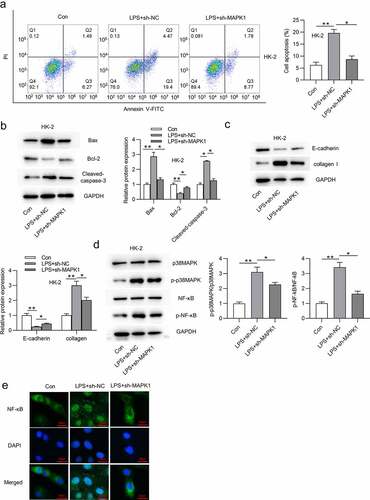
MAPK1 depletion alleviates sepsis-induced kidney injury in vivo
We then investigated the effect of MAPK1 on sepsis-induced AKI in vivo. First, the knockdown efficiency of MAPK1 is ensured in . The concentration of proinflammatory cytokines (TNF-α, IL-6 and IL-1β) was reduced by MAPK1 inhibition in model rats (). After silencing MAPK1, Bax and Cleaved caspase-3 protein levels were reduced while Bcl-2 protein level was enhanced in vivo compared with these in the Model+AAV-Sh-NC group (). Additionally, E-cadherin protein level was reduced while collagen I level was increased in Model group compared with those in sham group, and MAPK1 depletion partially reversed the alternations (). The distorted renal tubules, apoptotic or/and necrotic cell phenotypes were observed in Model group, but MAPK1 repression alleviated these effects (). In addition, serum creatinine levels in Model group were increased from 0.93 mg/dL to 1.89 mg/dL compared with those in the sham group, which indicated the successful establishment of the model of sepsis-induced AKI (). Moreover, increased levels of BUN, serum creatinine, and albumin-to-creatinine ratio in Model group were retarded after MAPK1 inhibition (). The These data suggested that MAPK1 deficiency alleviates sepsis-induced kidney injury in vivo.
Figure 3. MAPK1 depletion alleviates sepsis-induced kidney injury in vivo.
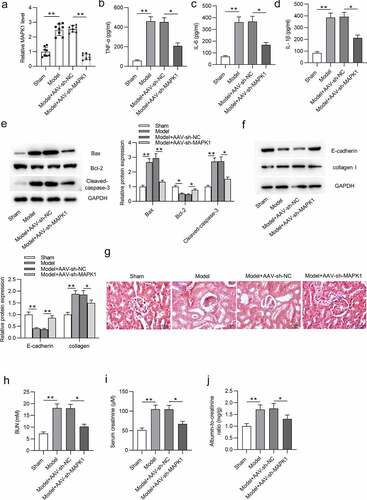
MiR-212-3p directly targets MAPK1
DIANA website (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=site%2Findex) was applied to search miRNAs that bind with MAPK1 with the screening condition of predicted score > 0.9. Eight candidate miRNAs are displayed in . Among these predicted miRNAs, miR-212-3p exhibited significantly low expression after LPS treatment (). Hence, miR-212-3p was selected for following study. The miR-212-3p level was effectively overexpressed by miR-212-3p mimics (). Then, the binding site between miR-212-3p and MAPK1 was predicted by Targetscan (http://www.targetscan.org/vert_72/) and was shown in . Additionally, the luciferase activity of MAPK1-WT reporters was damaged by miR-212-3p overexpression, but that of MAPK1-Mut reporters were not significantly changed (). Similarly, MAPK1 expression was also reduced by overexpressing miR-212-3p in LPS-induced HK-2 cells (). Next, miR-212-3p expression was silenced in LPS-treated HK-2 cells. RT-qPCR revealed a significant decrease in miR-212-3p expression after transfection of miR-212-3p inhibitor (). The luciferase activity of MAPK1 WT was increased in LPS-induced HK-2 cells transfected with miR-212-3p inhibitor (). In addition, mRNA expression of MAPK1 was upregulated by silenced miR-212-3p in LPS-treated HK-2 cells (). In summary, miR-212-3p directly targets MAPK1 3ʹ-UTR and negatively regulates MAPK1 in LPS-induced HK-2 cells.
Figure 4. MiR-212-3p directly targets MAPK1
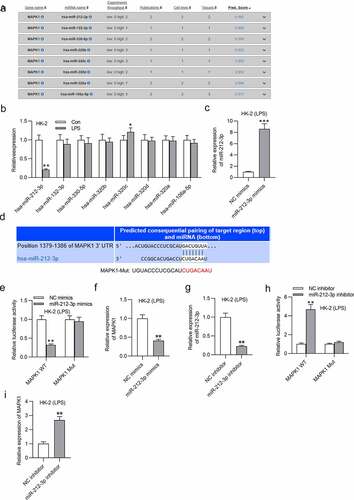
KCNQ1OT1 directly interacts with miR-212-3p
Many studies have reported that lncRNAs bind to miRNAs as a ceRNA and thus release mRNAs, and this ceRNA regulatory network is involved in the development of various human diseases [Citation63,Citation64]. Through the starBase website (http://starbase.sysu.edu.cn/), several lncRNAs are predicted to have binding site for miR-212-3p (Table S1). Particularly, KCNQ1OT1 and XIST possess more binding site on miR-21-3p than other candidate lncRNAs. RT-qPCR revealed that KCNQ1OT1 level was upregulated in the LPS group while XIST expression showed no significant changes (). The binding site between miR-212-3p and KCNQ1OT1 as well as the mutant sequence of KCNQ1OT1 were shown in . The luciferase activity of KCNQ1OT1-WT reporters was reduced by miR-212-3p mimics while that of KCNQ1OT1-Mut was not significantly affected (). KCNQ1OT1 and MAPK1 levels were both reduced by knockdown KCNQ1OT1 (). In conclusion, KCNQ1OT1 directly interacts with miR-212-3p.
Figure 5. KCNQ1OT1 directly interacts with miR-212-3p
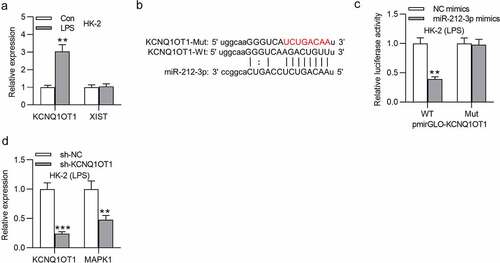
KCNQ1OT1 promotes inflammatory response in LPS-treated HK-2 cells via upregulation of MAPK1
As shown in , inhibition of KCNQ1OT1 decreased MAPK1 protein level, and this effect was rescued after transfection of pcDNA3.1/MAPK1. Furthermore, the decreased content of TNF-α, IL-6 and IL-1β caused by KCNQ1OT1 depletion could be reversed by MAPK1 overexpression (). These findings indicated that KCNQ1OT1 facilitates inflammatory response via upregulation of MAPK1.
Figure 6. KCNQ1OT1 promotes inflammatory response in LPS-treated HK-2 cells via upregulation of MAPK1
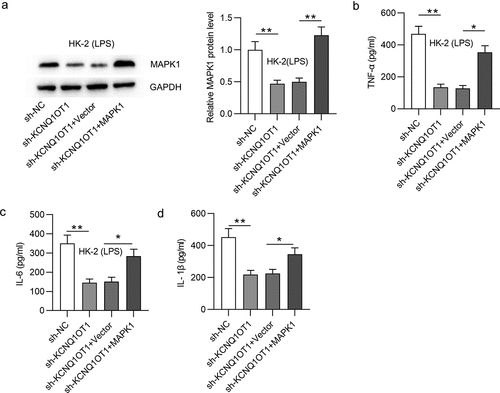
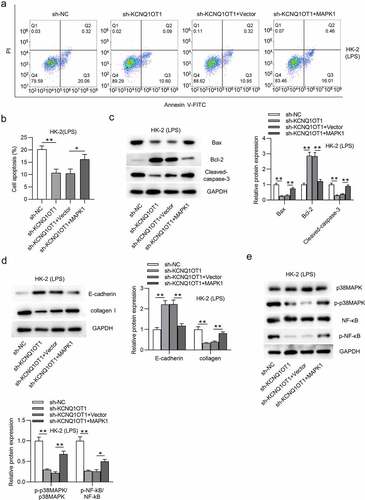
KCNQ1OT1 promotes HK-2 cell apoptosis and activates p38/NF-κB pathway via upregulation of MAPK1
Finally, we explored whether KCNQ1OT1 promotes HK-2 cell apoptosis and activates MAPK/NF-κB pathway by upregulation of MAPK1. Overexpression of MAPK1 counteracted the inhibitory effect on cell apoptosis caused by silencing KCNQ1OT1 (). The decrease in Bax and cleaved-caspase-3 protein levels and the increase in Bcl-2 protein level induced by silenced KCNQ1OT1 were reversed by MAPK1 up-regulation (). Additionally, MAPK1 overexpression reversed the enhancement of E-cadherin protein level and the reduction of collagen I protein level in LPS-treated HK-2 cells (). p-p38MAPK and p-NF-κB protein levels were reduced by suppressing KCNQ1OT1, but overexpressed MAPK1 neutralized this inhibitory effect (). These results revealed that KCNQ1OT1 promotes cell apoptosis and activates p38/NF-κB pathway via upregulation of MAPK1.
Figure 7. KCNQ1OT1 promotes HK-2 cell apoptosis and activates p38/NF-κB pathway in LPS-treated HK-2 cells via upregulation of MAPK1
Discussion
AKI is a heterogeneous group of conditions characterized by a sudden decrease in glomerular filtration rate (GFR) and then an increase in serum creatinine concentration (SCC) or oliguria [Citation65]. Clinically, three etiologies of AKI are prerenal, renal and postrenal one. Prerenal azotemia is featured with the decrease in GFR because of a decrease in renal perfusion pressure with little damage to renal parenchyma [Citation5]. Postrenal etiology of AKI is acute urinary tract obstruction which increases intratubular pressure and thereby decreases GFR [Citation5]. Renal causes of AKI are difficult to define, but evaluating damage to the tubules, glomeruli, interstitium and intrarenal blood vessels can be helpful for considering etiologies of intrinsic renal failure [Citation5]. In the present study, molecules involved in sepsis-induced AKI were investigated.
MAPK1 has been researched to be involved in the regulation of many diseases [Citation37,Citation38,Citation66]. However, the role of MAPK1 in sepsis-induced AKI remains unknown. MAPKs belong to the serine/threonine protein kinase family, and activated MAPKs stimulated cellular responses [Citation67,Citation68]. In this work, MAPK1 expression was upregulated in LPS-treated HK-2 cells. Silencing MAPK1 inhibited LPS-stimulated inflammatory response, HK-2 cell apoptosis, and EMT process. Additionally, MAPK1 depletion suppressed the protein level of renal fibrosis marker (collagen I) compared with the LPS group. NF-κB was reported to be a downstream target of p38-MAPK. Activation of p38-MAPK leads to phosphorylation of I-κB, which causes degradation of I-κB. NF-κB is depolymerized with I-κB and then activates nuclear translocation [Citation69,Citation70]. p38/NF-κB pathway plays an important role in many inflammatory diseases [Citation70–72]. In our study, silencing MAPK1 inactivated the p38/NF-κB pathway in LPS-induced HK-2 cells. Moreover, the impact of MAPK1 on sepsis-induced kidney injury in vivo was also revealed. After establishment of in vivo model of sepsis, serum creatinine levels show an average increase of 0.96 mg/dL in the model group. According to the criteria for the diagnosis of AKI, i.e., either 25% to 50% increase of baseline serum creatinine levels and/or an absolute elevation of 0.5–2.0 mg/dL from baseline, the results suggested that the establishment of sepsis-induced AKI model was achieved. The criterion was stipulated by the Acute Dialysis Quality Initiative (ADQI) proposing the RIFLE (Risk, Injury, Failure, Loss and End-stage kidney disease) system and Acute Kidney Injury Network (AKIN) [Citation73]. We found that silencing MAPK1 alleviated kidney injury in vivo, as shown by alleviation of inflammatory response, inhibition on cell apoptosis, and the decrease in levels of renal injury indicators (BUN, serum creatine) and albumin-to-creatinine ratio.
MiRNAs has been claimed to bind with mRNAs to promote degradation or inhibit translation [Citation74–76]. For example, miR-211 is downregulated in HK-2 cells under hypoxia-reoxygenation condition, which inhibits HK-2 cell apoptosis and alleviates ischemia/reperfusion-induced kidney injury by targeting TGFβR2/TGF-β/SMAD3 signals [Citation77]. Herein, miR-212-3p was predicted and confirmed to directly target MAPK1 3ʹ-UTR and negatively regulate MAPK1 expression. miR-212-3p was downregulated in LPS-treated HK-2 cells. miR-212-3p directly targeted 3ʹ-UTR of MAPK1 and negatively regulated MAPK1 expression in cells. Previously, miR-212-3p regulates HMGB1 to weaken LPS-induced inflammatory response in murine macrophages [Citation45]. Moreover, miR-212-3p decreased SOX5 expression to retard proliferation and strengthen apoptosis of fibroblast-like synoviocytes in rheumatoid arthritis [Citation78]. Hence, we hypothesized that miR-212-3p probably exert an anti-inflammatory effect on LPS-induced HK-2 cells.
Emerging studies proposed that MAPK1 was also regulated by certain lncRNAs in a competitive endogenous RNA (ceRNA) pattern [Citation79,Citation80]. For example, lncRNA metastasis associated lung adenocarcinoma transcript 1 (MALAT1) aggravates coronary atherosclerotic heart disease by interacting with miR-15b-5p to regulate MAPK1 and thus activating mTOR signaling pathway [Citation79]. Moreover, lncRNA taurine upregulated 1 (TUG1) was reported to exert a protective effect on lipopolysaccharide‑induced podocyte injury by competing with MAPK1 to interact with miR-197 [Citation80]. In our study, KCNQ1OT1 was validated to interact with miR-212-3p to upregulate MAPK1. According to previous studies, the role of KCNQ1OT1 is controversial in sepsis-associated diseases. In detail, KCNQ1OT1 displays low level in cardiac tissues of septic rats and alleviates myocardial injury [Citation54]. However, another study proposed that KCNQ1OT1 functioned as an activator of inflammatory response by modulating neutrophil extracellular trap formation in LPS-induced acute respiratory distress syndrome [Citation53]. Results from our work indicated that silencing KCNQ1OT1 restrained inflammatory response in LPS-stimulated HK-2 cells and inhibited cell apoptosis, and these results were reversed by MAPK1 overexpression. Additionally, KCNQ1OT1 was reported to participate in the activation of p38/NF-κB pathway in pituitary adenomas and acute myocardial injury [Citation81,Citation82]. Similarly, in our study, knockdown of KCNQ1OT1 inhibited the phosphorylation of p38 and NF-κB. However, MAPK1 overexpression reversed the suppressive effect of silenced KCNQ1OT1 on protein levels of phosphorylated p38MAPK and NF-κB. The results suggested that KCNQ1OT1 activates p38/NF-κB signaling by upregulating MAPK1.
In summary, KCNQ1OT1 aggravates sepsis-induced AKI by activating p38/NF-κB pathway via miR-212-3p/MAPK1 axis. This work may provide a promising biomarker for the treatment of sepsis-induced AKI. To some extent, there are limitations in our work. For example, the specific mechanism of LPS treatment inducing the dysregulation of KCNQ1OT1, miR-212-3p and MAPK1 in HK-2 cells remains unclear. In addition, the role of KCNQ1OT1 and miR-212-3p in sepsis-induced AKI needs to be further explored in the future.
Conclusion
KCNQ1OT1 is upregulated in LPS-stimulated HK-2 cells. KCNQ1OT1 deficiency inhibits inflammatory response in LPS-induced HK-2 cells and suppresses EMT, EndMT and cell apoptosis. KCNQ1OT1 interacts with miR-212-3p to regulate the expression of MAPK1. MAPK1 3ʹ-UTR is directly targeted by miR-212-3p. KCNQ1OT1 activates the p38/NF-κB signaling by upregulating MAPK1. MAPK1 depletion alleviates sepsis-induced kidney injury in vivo. In conclusion, KCNQ1OT1 competes with MAPK1 for the opportunity of interacting with miR-212-3p and release MAPK1 to activate p38/NF-κB signaling, thus aggravating sepsis-induced AKI.
Acknowledgements
We appreciate all participants for their contributions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data Availability Statement
The datasets used during the current study are available from the corresponding author on reasonable request.
Additional information
Funding
References
- Shum HP, Kong HH, Chan KC, et al. Septic acute kidney injury in critically ill patients - a single-center study on its incidence, clinical characteristics, and outcome predictors. Ren Fail. 2016 Jun;38(5):706–716.
- Gómez H, Kellum JA. Sepsis-induced acute kidney injury. Curr Opin Crit Care. 2016 Dec;22(6):546–553.
- Jörres A. Acute kidney injury. Minerva Urol Nefrol. 2016 Feb;68(1):47–48.
- Georgiadis G, Zisis IE, Docea AO, et al. Current concepts on the reno-protective effects of phosphodiesterase 5 inhibitors in acute kidney injury: systematic search and review. J Clin Med. 2020 Apr 29;9(5). DOI:10.3390/jcm9051284.
- Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol. 2012 Apr;2(2):1303–1353.
- Romagnoli S, Ricci Z, Ronco C. CRRT for sepsis-induced acute kidney injury. Curr Opin Crit Care. 2018 Dec;24(6):483–492.
- Manrique-Caballero CL, Del Rio-Pertuz G, Gomez H. Sepsis-associated acute kidney injury. Crit Care Clin. 2021 Apr;37(2):279–301.
- Cruz-Solbes AS, Youker K. Epithelial to mesenchymal transition (EMT) and endothelial to mesenchymal transition (EndMT): role and implications in kidney fibrosis. Results Probl Cell Differ. 2017;60:345–372.
- Lin JR, Zheng YJ, Zhang ZB, et al. Suppression of endothelial-to-mesenchymal transition by SIRT (Sirtuin) 3 alleviated the development of hypertensive renal injury. Hypertension. 2018 Aug;72(2):350–360.
- Tang TT, Lv LL, Wang B, et al. Employing macrophage-derived microvesicle for kidney-targeted delivery of dexamethasone: an Efficient therapeutic strategy against renal inflammation and fibrosis. Theranostics. 2019;9(16):4740–4755.
- Li J, Liu H, Srivastava SP, et al. Endothelial FGFR1 (Fibroblast Growth Factor Receptor 1) deficiency contributes differential fibrogenic effects in kidney and heart of diabetic mice. Hypertension. 2020 Dec;76(6):1935–1944.
- Peired A, Lazzeri E, Lasagni L, et al. Glomerular regeneration: when can the kidney regenerate from injury and what turns failure into success? Nephron Exp Nephrol. 2014;126(2):70.
- Poston JT, Koyner JL. Sepsis associated acute kidney injury. Bmj. 2019 Jan;9(364):k4891.
- Peerapornratana S, Manrique-Caballero CL, Gómez H, et al. Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019 Nov;96(5):1083–1099.
- Nandikanti DK, Gosmanova EO, Gosmanov AR. Acute kidney injury associated with linagliptin. Case Rep Endocrinol. 2016;2016:5695641.
- Chu C, Lu YP, Yin L, et al. The SGLT2 inhibitor empagliflozin might be a new approach for the prevention of acute kidney injury. Kidney Blood Press Res. 2019;44(2):149–157.
- Morigi M, Perico L, Rota C, et al. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest. 2015 Feb;125(2):715–726.
- Tsogbadrakh B, Ryu H, Ju KD, et al. AICAR, an AMPK activator, protects against cisplatin-induced acute kidney injury through the JAK/STAT/SOCS pathway. Biochem Biophys Res Commun. 2019 Feb 12;509(3):680–686.
- Tan C, Gu J, Li T, et al. Inhibition of aerobic glycolysis alleviates sepsis‑induced acute kidney injury by promoting lactate/Sirtuin 3/AMPK‑regulated autophagy. Int J Mol Med. 2021 Mar;47(3). doi:10.3892/ijmm.2021.4852.
- Camin RM, Cols M, Chevarria JL, et al. Acute kidney injury secondary to a combination of renin-angiotensin system inhibitors, diuretics and NSAIDS: “the triple whammy”. Nefrologia. 2015;35(2):197–206.
- Giani JF, Veiras LC, Shen JZY, et al. Novel roles of the renal angiotensin-converting enzyme. Mol Cell Endocrinol. 2021 Jun;529:111257.
- Chvojka J, Sýkora R, Karvunidis T, et al. New developments in septic acute kidney injury. Physiol Res. 2010;59(6):859–869.
- Kane-Gill SL, Meersch M, Bell M. Biomarker-guided management of acute kidney injury. Curr Opin Crit Care. 2020 Dec;26(6):556–562.
- Li M, Li CM, Ye ZC, et al. Sirt3 modulates fatty acid oxidation and attenuates cisplatin-induced AKI in mice. J Cell Mol Med. 2020 May;24(9):5109–5121.
- Zhu P, Ren M, Yang C, et al. Involvement of RAGE, MAPK and NF-κB pathways in AGEs-induced MMP-9 activation in HaCaT keratinocytes. Exp Dermatol. 2012 Feb;21(2):123–129.
- Zyuz’kov GN, Zhdanov VV, Miroshnichenko LA, et al. Involvement of PI3K, MAPK ERK1/2 and p38 in functional stimulation of mesenchymal progenitor cells by alkaloid songorine. Bull Exp Biol Med. 2015 May;159(1):58–61.
- Zou Y, Zeng S, Huang M, et al. Inhibition of 6-phosphofructo-2-kinase suppresses fibroblast-like synoviocytes-mediated synovial inflammation and joint destruction in rheumatoid arthritis. Br J Pharmacol. 2017 May;174(9):893–908.
- Zyuz’kov GN, Zhdanov VV, Udut EV, et al. Peculiarities of intracellular signal transduction in the regulation of functions of mesenchymal, neural, and hematopoietic progenitor cells. Bull Exp Biol Med. 2019 Jun;167(2):201–206.
- Zhou QM, Chen QL, Du J, et al. Synergistic effect of combinatorial treatment with curcumin and mitomycin C on the induction of apoptosis of breast cancer cells: a cDNA microarray analysis. Int J Mol Sci. 2014 Sep 15;15(9):16284–16301.
- Zhou X, Wang C, Tian J, et al. Mitogen-activated protein kinase mediates mevalonate-stimulated human mesangial cell proliferation. Mol Med Rep. 2015 Aug;12(2):2643–2649.
- Zyromski N, Murr MM. Evolving concepts in the pathophysiology of acute pancreatitis. Surgery. 2003 Mar;133(3):235–237.
- Zyuz’kov GN, Miroshnichenko LA, Polyakova TY, et al. Role of MAPK ERK1/2 and p38 in the realization of growth potential of various types of regeneration-competent cells in mouse neural tissue during ethanol-induced neurodegeneration in vitro. Bull Exp Biol Med. 2019 Jun;167(2):229–232.
- Zu L, He J, Jiang H, et al. Bacterial endotoxin stimulates adipose lipolysis via toll-like receptor 4 and extracellular signal-regulated kinase pathway. J Biol Chem. 2009 Feb 27;284(9):5915–5926.
- Zula JA, Green HC, Ransohoff RM, et al. The role of cell type-specific responses in IFN-β therapy of multiple sclerosis. Proc Natl Acad Sci U S A. 2011 Dec 6;108(49):19689–19694.
- Zuo J, Dou DY, Wang HF, et al. Reactive oxygen species mediated NF-κB/p38 feedback loop implicated in proliferation inhibition of HFLS-RA cells induced by 1,7-dihydroxy-3,4-dimethoxyxanthone. Biomed Pharmacother. 2017 Oct;94:1002–1009.
- Zuo Q, Shi M, Chen J, et al. The Ras signaling pathway mediates cetuximab resistance in nasopharyngeal carcinoma. Biomed Pharmacother. 2011 Jun;65(3):168–174.
- Zhou CL, Li F, Wu XW, et al. Overexpression of miRNA-433-5p protects acute spinal cord injury through activating MAPK1. Eur Rev Med Pharmacol Sci. 2020 Mar;24(6):2829–2835.
- Zhu S, Song W, Sun Y, et al. MiR-342 attenuates lipopolysaccharide-induced acute lung injury via inhibiting MAPK1 expression. Clin Exp Pharmacol Physiol. 2020 Aug;47(8):1448–1454.
- Guo F, Xing Y, Zhou Z, et al. Guanine-nucleotide exchange factor H1 mediates lipopolysaccharide-induced interleukin 6 and tumor necrosis factor α expression in endothelial cells via activation of nuclear factor κB. Shock. 2012 May;37(5):531–538.
- Tafrihi M, Hasheminasab E. MiRNAs: biology, biogenesis, their web-based tools, and databases. Microrna. 2019;8(1):4–27.
- Liu B, Li J, Cairns MJ. Identifying miRNAs, targets and functions. Brief Bioinform. 2014 Jan;15(1):1–19.
- Srivastava SP, Koya D, Kanasaki K. MicroRNAs in kidney fibrosis and diabetic nephropathy: roles on EMT and EndMT. Biomed Res Int. 2013;2013:125469.
- Srivastava SP, Hedayat AF, Kanasaki K, et al. microRNA crosstalk influences epithelial-to-mesenchymal, endothelial-to-mesenchymal, and macrophage-to-mesenchymal transitions in the kidney. Front Pharmacol. 2019;10:904.
- Wang X, Wang Y, and Kong M, et al. MiR-22-3p suppresses sepsis-induced acute kidney injury by targeting PTEN. Biosci Rep. 2020 Jun 26;40(6): BSR20200527.
- Chen W, Ma X, Zhang P, et al. MiR-212-3p inhibits LPS-induced inflammatory response through targeting HMGB1 in murine macrophages. Exp Cell Res. 2017 Jan 15;350(2):318–326.
- Zhu S, Wang J, He Y, et al. Peptides/proteins encoded by non-coding RNA: a novel resource bank for drug targets and biomarkers. Front Pharmacol. 2018;9:1295.
- Zong W, Ju S, Jing R, et al. Long non-coding RNA-mediated regulation of signaling pathways in gastric cancer. Clin Chem Lab Med. 2018 Oct 25;56(11):1828–1837.
- Zur Bruegge J, Einspanier R, Sharbati S, et al. Ahead: long non-coding RNAs in bacterial infections. Front Cell Infect Microbiol. 2017;7:95.
- Wang Y, Tan J, Xu C, et al. Identification and construction of lncRNA-associated ceRNA network in diabetic kidney disease. Medicine (Baltimore). 2021 Jun 4;100(22):e26062.
- Liu X, Zhu N, Zhang B, et al. Long noncoding RNA TCONS_00016406 attenuates lipopolysaccharide-induced acute kidney injury by regulating the miR-687/PTEN pathway. Front Physiol. 2020;11:622.
- Yang N, Wang H, Zhang L, et al. Long non-coding RNA SNHG14 aggravates LPS-induced acute kidney injury through regulating miR-495-3p/HIPK1. Acta Biochim Biophys Sin (Shanghai). 2021 May 21;53(6):719–728.
- Chen FR, Zheng LM, Wu DC, et al. [Regulatory relationship between lncRNA KCNQ1OT1 and miR-146a-3p in preeclampsia]. Zhonghua Fu Chan Ke Za Zhi. 2020 Aug 25;55(8):535–543.
- Jiang X, Yu M, Zhu T, et al. Kcnq1ot1/miR-381-3p/ETS2 axis regulates inflammation in mouse models of acute respiratory distress syndrome. Mol Ther Nucleic Acids. 2020 Mar 6;19:179–189.
- Sun F, Yuan W, Wu H, et al. LncRNA KCNQ1OT1 attenuates sepsis-induced myocardial injury via regulating miR-192-5p/XIAP axis. Exp Biol Med (Maywood). 2020 Apr;245(7):620–630.
- Jiang ZJ, Zhang MY, Fan ZW, et al. Influence of lncRNA HOTAIR on acute kidney injury in sepsis rats through regulating miR-34a/Bcl-2 pathway. Eur Rev Med Pharmacol Sci. 2019 Apr;23(8):3512–3519.
- Rocca CJ, Ur SN, Harrison F, et al. rAAV9 combined with renal vein injection is optimal for kidney-targeted gene delivery: conclusion of a comparative study. Gene Ther. 2014 Jun;21(6):618–628.
- Wang J, Song J, Li Y, et al. Down-regulation of LncRNA CRNDE aggravates kidney injury via increasing MiR-181a-5p in sepsis. Int Immunopharmacol. 2020 Feb;79:105933.
- Lu S, Wu H, Xu J, et al. SIKIAT1/miR-96/FOXA1 axis regulates sepsis-induced kidney injury through induction of apoptosis. Inflamm Res. 2020 Jul;69(7):645–656.
- Listwak SJ, Rathore P, Herkenham M. Minimal NF-κB activity in neurons. Neuroscience. 2013 10;Oct(250):282–299.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001 Dec;25(4):402–408.
- Gallo-Oller G, Ordoñez R, Dotor J. A new background subtraction method for Western blot densitometry band quantification through image analysis software. J Immunol Methods. 2018 Jun;457:1–5.
- Park E, Cho M, Ki CS. Correct use of repeated measures analysis of variance. Korean J Lab Med. 2009 Feb;29(1):1–9.
- Li Y, Huo C, Lin X, et al. Computational identification of cross-talking ceRNAs. Adv Exp Med Biol. 2018;1094:97–108.
- Qi X, Zhang DH, Wu N, et al. ceRNA in cancer: possible functions and clinical implications. J Med Genet. 2015 Oct;52(10):710–718.
- Levey AS, James MT. Acute kidney injury. Ann Intern Med. 2017 Nov 7;167(9):Itc66–itc80.
- Li XW, Tuergan M, Abulizi G. Expression of MAPK1 in cervical cancer and effect of MAPK1 gene silencing on epithelial-mesenchymal transition, invasion and metastasis. Asian Pac J Trop Med. 2015 Nov;8(11):937–943.
- Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010 Apr;1802(4):396–405.
- Zhang WB, Yang F, Wang Y, et al. Inhibition of HDAC6 attenuates LPS-induced inflammation in macrophages by regulating oxidative stress and suppressing the TLR4-MAPK/NF-κB pathways. Biomed Pharmacother. 2019 Sep;117:109166.
- Verma VK, Malik S, Narayanan SP, et al. Role of MAPK/NF-κB pathway in cardioprotective effect of Morin in isoproterenol induced myocardial injury in rats. Mol Biol Rep. 2019 Feb;46(1):1139–1148.
- Alam S, Liu Q, Liu S, et al. Up-regulated cathepsin C induces macrophage M1 polarization through FAK-triggered p38 MAPK/NF-κB pathway. Exp Cell Res. 2019 Sep 15;382(2):111472.
- Sun P, Zhou K, Wang S, et al. Involvement of MAPK/NF-κB signaling in the activation of the cholinergic anti-inflammatory pathway in experimental colitis by chronic vagus nerve stimulation. PLoS One. 2013;8(8):e69424.
- Yu X, Cui L, Hou F, et al. Angiotensin-converting enzyme 2-angiotensin (1-7)-Mas axis prevents pancreatic acinar cell inflammatory response via inhibition of the p38 mitogen-activated protein kinase/nuclear factor-κB pathway. Int J Mol Med. 2018 Jan;41(1):409–420.
- Mamoulakis C, Tsarouhas K, Fragkiadoulaki I, et al. Contrast-induced nephropathy: basic concepts, pathophysiological implications and prevention strategies. Pharmacol Ther. 2017 Dec;180:99–112.
- Yu L, Yang J. MiR-29 increases apoptosis of retinal ganglion cells in rat glaucoma model through AKT signaling pathway. Panminerva Med. 2019 Jun 28;63:90–91.
- Wang Y, Hou L, Yuan X, et al. miR-483-3p promotes cell proliferation and suppresses apoptosis in rheumatoid arthritis fibroblast-like synoviocytes by targeting IGF-1. Biomed Pharmacother. 2020 Jul 15;130:110519.
- Chai Z, Gong J, Zheng P, et al. Inhibition of miR-19a-3p decreases cerebral ischemia/reperfusion injury by targeting IGFBP3 in vivo and in vitro. Biol Res. 2020 Apr 20;53(1):17.
- Shang J, Sun S, Zhang L, et al. miR-211 alleviates ischaemia/reperfusion-induced kidney injury by targeting TGFβR2/TGF-β/SMAD3 pathway. Bioengineered. 2020 Dec;11(1):547–557.
- Liu Y, Zhang XL, Li XF, et al. miR-212-3p reduced proliferation, and promoted apoptosis of fibroblast-like synoviocytes via down-regulating SOX5 in rheumatoid arthritis. Eur Rev Med Pharmacol Sci. 2018 Jan;22(2):461–471.
- Zhu Y, Yang T, Duan J, et al. MALAT1/miR-15b-5p/MAPK1 mediates endothelial progenitor cells autophagy and affects coronary atherosclerotic heart disease via mTOR signaling pathway. Aging (Albany NY). 2019 Feb 21;11(4):1089–1109.
- Zhao D, Liu Z, Zhang H. The protective effect of the TUG1/miR‑197/MAPK1 axis on lipopolysaccharide‑induced podocyte injury. Mol Med Rep. 2019 Jul;20(1):49–56.
- Li X, Dai Y, Yan S, et al. Down-regulation of lncRNA KCNQ1OT1 protects against myocardial ischemia/reperfusion injury following acute myocardial infarction. Biochem Biophys Res Commun. 2017 Sep 30;491(4):1026–1033.
- Xue YH, Ge YQ. Construction of lncRNA regulatory networks reveal the key lncRNAs associated with Pituitary adenomas progression. Math Biosci Eng. 2020 Jan 6;17(3):2138–2149.