
ABSTRACT
MiRNAs (microRNAs) participate in colorectal cancer (CRC) progression and act as potential biomarkers for CRC prognosis. In this study, we investigated the mechanisms of microRNA-92a (miR-92a) in CRC. Expressions of miR-92a and SOCS3 (Suppressor Of Cytokine Signaling 3) were investigated by qRT-PCR in CRC cell lines and 30 cases of CRC. The self-renewal capacity and proliferation of CRC stem cells were estimated by the sphere formation assay, EdU staining, and Flow cytometry analysis. Moreover, the interplay between miR-92a and SOCS3 in CRC cells was validated by luciferase reporter experiments. MiR-92a was found to be remarkably increased while SOCS3 was significantly downregulated in CRC tissues. Inhibition of miR-92a or SOCS3 attenuated the sphere formation capacity, decreased expressions of stemness-related proteins, and inhibited the proliferation of cancer stem-like cells. Knockdown of SOCS3 reversed the repressive impacts of miR-92a inhibitors on self-renewal and growth of CRC cancer stem cells. This study suggested that miR-92a functions as an oncogene of CRC through mediating the stemness of colorectal cancer cells by directly binding and repressing SOCS3.
Abbreviations
CRC, cancer cell; CSCs, cancer stem cells; SOCS3, suppressor of cytokine signaling 3.
Introduction
Colorectal cancer (CRC), also termed bowel cancer, is a type of tumor that arises from the colon or rectum [Citation1]. The number of CRC patients has continued to rise in recent years, and currently it ranks as the fourth most frequent tumor type in the United States and as the second most fatal malignancy [Citation2]. In 2019, estimated 776,120 males and 768,650 females will be diagnosed as CRC in the United States [Citation3]. Accumulating shreds of evidence have documented that cancer stem cells (CSCs), a subset of cancer cells with strong self-renewal and multiline-differentiation abilities, might act as an important regulator in tumor initiation, development, metastasis, relapse, and drug resistance. Therefore, CSCs are considered to be one of the major hurdles for tumor eradication [Citation4,Citation5]. Understanding the molecular mechanisms and signal cascades initiating CSCs will contribute to developing more effective diagnostic and therapeutic strategies for clinical treatment. Compelling evidence has established that CSCs play a key role during the tumorigenesis of CRC [Citation6]. Nevertheless, the critical factors that determine the acquisition and maintenance of CRC CSCs characters remain unclear.
MicroRNAs (miRNAs), as a type of small non-coding RNAs, are widely presented in mammal cells [Citation7]. Studies reported that miRNAs are closely associated with multiple critical biological processes, such as epigenetic molecular regulation, DNA repair, cell proliferation, apoptosis, and signal transduction [Citation8]. Abnormal expressions of miRNA lead to the destruction of normal physiological activities in mammal cells, resulting in various human diseases, including neurodegenerative diseases, metabolic diseases, autoimmune diseases, and cancers [Citation9,Citation10]. The implication of miRNAs in CRC has been well established by numerous studies showing that miRNAs can serve as both oncogene and tumor repressors of CRC through diverse mechanisms [Citation11,Citation12]. In recent years, miRNAs have been reported to affect CRC tumor growth by regulating the property of CRC CSCs, implying that miRNAs might be a potential regulator of CRC stemness acquisition and maintenance [Citation13]. MiR-92a is a critical miRNA that has been correlated with multiple cancer types, such as esophageal squamous cell cancer, hepatocellular carcinoma, and glioma [Citation14–16]. MiR-92a was recently reported by Peng Q et al. as a promising molecule in predicting risk, recurrence, and survival of CRC [Citation17]. However, the function of miR-92a in CRC stemness has not been determined.
SOCS3 (suppressor of cytokine signaling 3), a critical subtype of the SOCS family, is a negative modulatory agent in growth factors and cytokines-related signaling cascades [Citation18,Citation19]. SOCS3 can repress the JAK-STAT pathway, inhibit cancer cell malignant transformation, and facilitate cancer cell apoptosis [Citation20]. Thus, activation of SOCS3 has been considered a promising therapeutic treatment of tumors [Citation21]. The role of SOCS3 in CRC was demonstrated by previous studies showing that SOCS3 overexpression represses CRC cell proliferation and invasion while facilitating CRC cell apoptosis [Citation22]. The interaction between SOCS3 and miRNAs plays a critical role in CRC tumor progression [Citation23].
The aim of this study was to investigate whether miR-92a and SOCS3 play a role in the stemness of CRC cells. We hypothesized that miR-92a promotes stem-like property of CRC by targeting SOCS3, and thus deteriorates progress of CRC. We also investigated the biological functions of miR-92a and SOCS3 in CRC stemness in vitro, attempting to further explore the mechanisms of CRC.
Materials and methods
CRC tumor collection and cell culture
Thirty CRC tissue and matched non-tumor tissues were obtained from patients who were diagnosed at the Second Affiliated Hospital of North Sichuan Medical College as CRC and without chemotherapy. Specimens were kept at −80°C until the study began. Every subject received a written informed consent before this study. This study was approved by the Ethics Committee of the Second Affiliated Hospital of North Sichuan Medical College. Four human CRC cell lines (LoVo, SW480, HT29, and HCT116) and one normal colon epithelial cell line (FHC) were offered by the Chinese Academy of Science cell bank (Shanghai, China). Cells were maintained in RPMI-1640 medium (Invitrogen, USA) supplemented with 10% fetal bovine serum (Gibco, USA).
Quantitative real-time PCR (qRT-PCR) assay
Total RNAs were extracted from CRC tissues and cells using the TRIzol reagent (Invitrogen) according to the manufacturers’ instructions. The quality of RNAs was tested via NanoDro 2000 c (Thermo Scientific, USA). Subsequently, 3 μg RNAs were utilized as templates to produce cDNA using PrimeScript RT Reagent (Takara, Dalian, China). The SOCS3 mRNA and miR-92a were detected by a SYBR Premix Taq (Takara). The PCR processes were completed on an ABI 7300 Real-Time system (Ambion, USA) with parameters of 95°C for 40 s, 40 cycles of 95°C for 25 s, 58°C for 40 s, and 72°C for 55 s. Primers are shown in .
Table 1. The sequences of primers for Realtime PCR
Western blot assay
Total proteins were extracted from CRC tissue and cell samples by the RIPA reagent obtained from Sigma, USA, and separated using 10% SDS-PAGE. The isolated proteins were transferred into PVDF membranes followed by 5% nonfat milk incubation to minimize the noise. Next, the membranes were immersed in milk containing primary antibodies against SOCS3 (Rabbit, 1:2000, ab16030, Abcam, UK), CD133 (Rabbit, 1:1000, ab216323, Abcam), SOX2 (Mouse, 1:2000, ab171380, Abcam), OCT4 (Rabbit, 1:5000, ab109183, Abcam), AKT (Rabbit, 1:500, ab8805, Abcam), STAT3 (Mouse, 1:5000, ab119352, Abcam), p-AKT (Rabbit, 1:1000, ab38449, Abcam), p-STAT3 (Rabbit, 1:5000, ab76315, Abcam), and GAPDH (Rabbit, 1:3000, ab124905, Abcam), and incubated overnight. After excess primary antibodies were washed off by PBS, the membranes were probed by HRP-conjunct secondary antibodies, and the signals were visualized using the ECL reagent (Bio-Rad).
RNA transfection
The miR-92a inhibitors, pcDNA3.0-SOCS3, SOCS3 siRNA, and their corresponding controls, including the miR-92a negative control and SOCS3 negative control, were provided by GenePharma (Shanghai, China). For cell transfection, CRC cells (2 × 106 cells/well) were seeded into 96-well plates and cultured at 37°C for 10 h. Cells were then transfected with the indicated RNAs using Lipofectamine 3000 (Invitrogen).
CSC cell culture and sphere formation analysis
CRC cancer stem-like cells (CSCs) were enriched using a serum-free culture medium. CRC CSCs were maintained in ultra-low attachment flasks (Corning, MA, USA) as spheres in RPMI-1640 containing nonessential amino acids (Fisher, USA), penicillin–streptomycin (Fisher), B27 supplement (Invitrogen, USA), and epidermal growth factor (50 ng/mL, Invitrogen). For sphere formation detection, CSCs (1,000 cells) were seeded into 6-well ultra-low attachment plates (Corning), and tumor spheres were counted after 2 weeks of culture using a microscope (Olympus).
EdU staining
The Cell-Light™ EdU detection Kit (Ribobio) was used to examine cell proliferation. In brief, CRC cells were plated into poly-L-lysine coated 96-well plates and maintained at 37°C for 8 h. Next, EdU (50 μM) was added to each well and allowed to incubate with CSC cells for an additional 24 h, followed by fixation of 4% paraformaldehyde. Finally, the treated cells were observed by a DMR fluorescent microscope (Leica, Germany).
Luciferase reporter assay
To validate the interplay between SOCS3 and miR-92a, a luciferase reporter assay was conducted on CRC cells. Briefly, the wild-type miR-92a target region of SOCS3 3ʹ-UTR was cloned into psi-CHECK2 (Origene, Rockville, MD) to establish a luciferase reporter plasmid, SOCS3-WT. The mutant target region was also inserted into psi-CHECK2 to construct SOCS3-Mut. The SOCS3-WT or SOCS3-Mut was co-transfected into CRC cells with miR-92a mimics or inhibitors using Lipofectamine 3000 (Invitrogen). The change of luciferase activity was monitored using a Dual-Luciferase Assay System (Promega) kit under the guidance of the manufacturer.
Statistical analysis
All data are shown as mean ± SEM, and one-way ANOVA analysis, or student’s t test, was conducted on the GraphPad Prism (Version 7.0, USA) to estimate the difference between groups. P < 0.05 was regarded as statistically significant.
Results
In this study, we first detected the differential expression of SOCS3 in cancer tissues and para-cancerous tissues, and then detected the expression level of miR-92a in tumor cell lines. Then, in the colorectal cancer cell line, miR-92a was knocked down, and then the cell sphere formation, the expression level of CD133 et al. and cell proliferation activity were detected. Then, the 3ʹUTR region in which miR-92a can target SOCS3 was detected by dual-Luciferase report assay, and the inhibitory effect of miR-92a on the expression of SOCS3 was determined by Western blot. In colorectal cancer cell line, SOCS3 was over-expressed, and the cell sphere formation, the expression level of CD133 et al. and cell proliferation activity were detected. Finally, knockdown of miR-92a and SOCS3, cell sphere formation, expression level of CD133 et al. and cell proliferation activity of colorectal cancer cells were detected. Experiments above elucidate the effect of miR-92a/SOCS3 on the malignant behavior of colorectal cancer cells.
SOCS3 was lowly expressed while miR-92a was highly expressed in CRC
To explore the role of SOCS3 and miR-92a during CRC tumorigenesis, we first examined expressions of SOCS3 in 30 CRC clinical tissues and cell lines. Results obtained from qRT-PCR showed that SOCS3 mRNA was remarkably decreased in CRC tissue samples compared with matched normal samples (, ***P < 0.001). Moreover, protein expression of SOCS3 was also downregulated in CRC tumor samples compared to that in non-tumor samples (). Results of qRT-PCR showed that expression of SOCS3 in cancer cell lines was lower than that in FHC cells (). Further experiments presented that miR-92a expression was higher in tumor tissues than non-tumor tissues (). In addition, miR-92a was upregulated in CRC cell lines (LoVo, SW480, HT29, and HCT116) compared to the FHC cell line (, **P < 0.01, ***P < 0.001). We also found that miR-92a expression in CRC cell lines, LoVo and SW480 was higher than that in HT29 and HCT116. Therefore, CRC cell lines, LoVo and SW480 were used in subsequent experiments.
Figure 1. SOCS3 was lowly expressed while miR-92a was highly expressed in CRC. (a) The SOCS3 mRNA level was examined by qRT-PCR in CRC and matched normal samples, ***P < 0.001 vs non-tumor samples. (b) Expression of the SOCS3 protein was measured using Western blot in CRC and matched normal samples, *P < 0.05 vs non-tumor samples. (c) Expression of SOCS3 was measured using qRT-PCR in FHC and CRC cell lines. * P < 0.05, **P < 0.01, ***P < 0.001 vs FHC group. (d) Expression of miR-92a was measured using qRT-PCR in clinical tissues. (e) The relative levels of miR-92a in four CRC cell lines and FHC cells were detected by qRT-PCR, **P < 0.01, ***P < 0.001 vs FHC group.
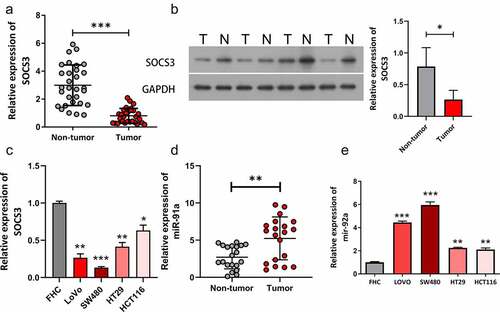
Inhibition of miR-92a repressed self-renewal and growth of CSC cells
Due to that tumor stemness has been closely correlated to tumor initiation, migration, recurrence, and even drug resistance, we then investigated the effect of miR-92a on the stemness-related properties of CRC CSC cells, including the self-renewal capacity and levels of CRC CSC markers. By using two CRC CSC cell lines (SW480 and LoVo) treated with the miR-92a inhibitor or matched inhibitor NC, we found that miR-92a inhibition markedly repressed the sphere formation ability (, **P < 0.01). To further confirm this finding, qRT-PCR and Western blot analysis were used to estimate expressions of stemness-related proteins in SW480 and LoVo CSC cells. Results indicated that CD133, SOX2, and OCT4 were all significantly elevated in CSC cells than in non-CSC CRC cells, and knockdown of miR-92a remarkably decreased their expression in CSC cells (, **P < 0.01, ***P < 0.001; ## P < 0.01). We performed EdU staining to evaluate the role of miR-92a inhibition on CRC CSCs growth. Results from EdU staining revealed that miR-92a inhibition resulted in significant repression of cell growth (). These results demonstrated that miR-92a inhibition remarkably suppressed self-renewal and growth of CSC cells.
Figure 2. Inhibition of miR-92a repressed self-renewal and growth of CSC cells. (a) A serial sphere propagation experiment was used to estimate the influence of miR-92a knockdown on CSC self-renewal, **P < 0.01 vs CSC+NC. (b) Levels of CD133, Sox2, and Oct4 were evaluated by qRT-PCR in SW480 and LoVo cells, **P < 0.01, ***P < 0.001 vs blank; ## P < 0.01 vs CSC+NC. (c) Protein expressions of CD133, SOX2, and OCT4 were detected in SW480 and LoVo cells. (d) EdU staining was used to assess the effects of miR-92a knockdown on CSC cell proliferation.
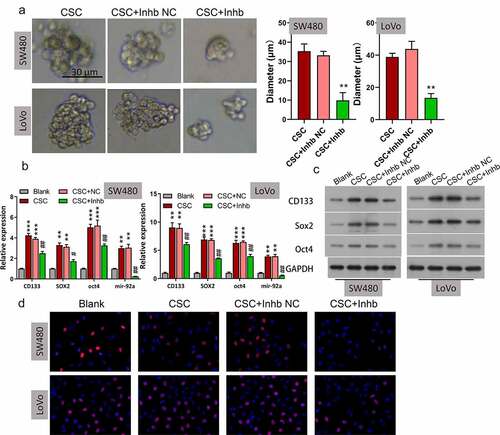
SOCS3 was bound and negatively modulated by miR-92a in CSC
By using bioinformatics analysis, it was found that the 3ʹ-UTR regions of SOCS3 possessed a miR-92a binding sequence (). We investigated whether there is an interaction between miR-92a and SOCS3 in CRC through dual-luciferase reporter experiments. The luciferase activities of CRC cells driven by the wild-type SOCS3 plasmid could be markedly enhanced or attenuated by the miR-92a inhibitor or miR-92a mimics, respectively. In contrast, the luciferase activities of those cells driven by the mutant type of the SOCS3 plasmid could not be affected by the miR-92 inhibitor or mimics (, *P < 0.05, **P < 0.01). In addition, using Western blot, it was demonstrated that miR-92a overexpression resulted in a remarkable downregulation of SOCS3 in SW480 and LoVo cells (). These findings indicated that SOCS3 was bound and negatively modulated by miR-92a.
Figure 3. SOCS3 was bound and negatively modulated by miR-92a in CSC. (a) The putative binding site sequence between miR-92a and SOC3C 3ʹ-UTR. (b) Interplay between miR-92a and SOCS3 was validated by luciferase reporter experiments, *P < 0.05 vs inhibitor NC, *P < 0.01 vs mimics NC. (c) Western blot analysis of SOCS3 in miR-92a silenced SW480 and LoVo cells.
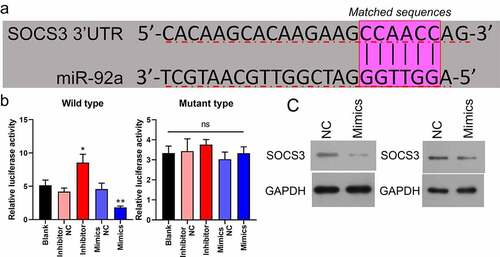
Overexpression of SOCS3 repressed self-renewal and growth of CSC cells
To understand whether SOCS3 mediates self-renewal and growth of CRC in CSC cells, SOCS3 was overexpressed in SW480 and LoVo CSC cells, and then the overexpression efficiency of SOCS3 was examined. Results indicated that SOCS3 was significantly increased in SOCS3 transfected SW480 and LoVo CSC cells (). In the sphere formation analysis, it was found that SOCS3 overexpression markedly attenuated the sphere formation capacity of SW480 and LoVo CSC cells (, **P < 0.01). Also, it was found that CD133, SOX2, and OCT4 expression was downregulated in SOCS3 transfected SW480 and LoVo CSC cells compared to negative control groups (, ***P < 0.001, #P < 0.05). Moreover, results obtained from EdU staining indicated that SOCS3 overexpression caused a significant repression of cell proliferation of SW480 and LoVo CSC cells (, ***P < 0.001). In addition, we found that the protein levels of p-AKT and p-STAT3 were remarkably downregulated in SOCS3 treated SW480 and LoVo CSC cells compared to negative control cells (). These results demonstrated that overexpression of SOCS3 repressed self-renewal and growth of CSC cells.
Figure 4. Overexpression of SOCS3 repressed self-renewal and growth of CSC cells. (a and b) qRT-PCR and Western blot were used to examine the SOCS3 overexpression efficiency in SW480 and LoVo cells, ***P < 0.001 vs NC. (c) Influences of SOCS3 overexpression on CSC self-renewal were estimated by the sphere propagation assay in SW480 and LoVo cells, **P < 0.01 vs CSC+NC. (d) qRT-PCR and (e) Western blot were carried out in SOCS3 overexpressed SW480 and LoVo CSC cells to examine the protein levels of CD133, SOX2, and OXT4, ***P < 0.001 vs blank; ##P < 0.01 vs CSC+NC. (f) Cell proliferation was evaluated by EdU staining in SW480 and LoVo CSC. (g) Protein expressions of SOCS3, AKT/p-AKT, and STAT3/p-STAT3 in SOCS3 overexpressed SW480 and LoVo CSC cells were assessed via Western blot.
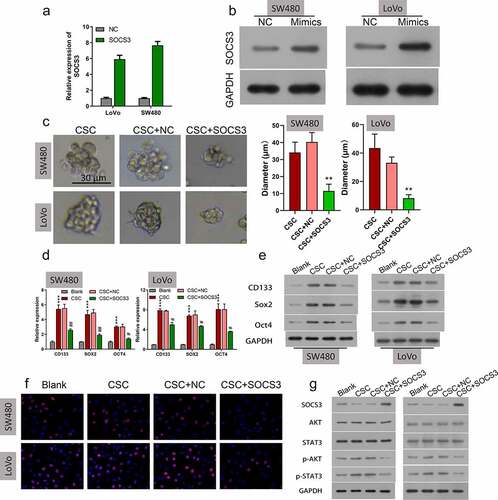
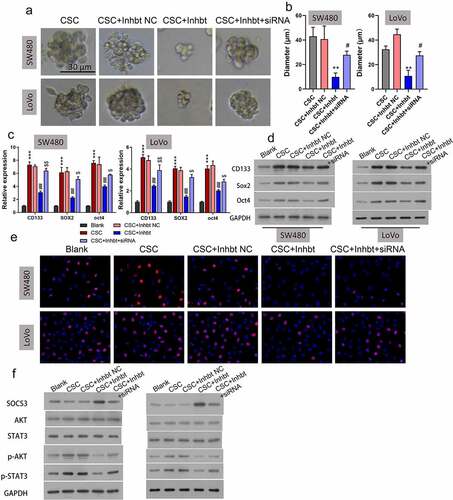
Knockdown of SOCS3 reversed the repressive effects of the miR-92a inhibitor on self-renewal and growth of CSC cells
Because miR-92a inhibition and SOCS3 overexpression were both demonstrated to repress the self-renewal and growth of CRC CSC cells, and miR-92a could bind and negatively regulate SOSC3, we speculated that SOCS3 might be involved in the tumorigenesis of CRC by acting downstream of miR-92a. To validate this speculation, we treated miR-92a silenced SW480 and LoVo CSC cells with SOCS siRNA, followed by the detection of cell self-renewal and growth. Results illustrated that treatment of SOSC3 siRNA could remarkably reverse the repression of sphere formation ability induced by the miR-92a inhibitor (, **P < 0.01, #P < 0.05). Correspondingly, the miR-92a inhibitor caused downregulation of CD133, SOX2, and OCT4 that was also demonstrated to be abolished by SOCS3 siRNA treatment in SW480 and LoVo CSC cells at both mRNA (, ***P < 0.001, ##P < 0.01) and protein () levels. We also demonstrated that the repression of cell proliferation of SW480 and LoVo CSC cells caused by miR-92a inhibitor transfection could be reversed by SOCS3 siRNA (). In addition, the reduction of p-AKT and p-STAT3 in miR-92a silenced SW480 and LoVo CSC cells were also abrogated by the SOCS3 siRNA treatment (). These findings indicated that knockdown of SOCS3 reversed the repressive effects of the miR-92a inhibitor on self-renewal and growth of CSC cells.
Figure 5. Knockdown of SOCS3 reduced the repressive functions of the miR-92a inhibitor on self-renewal and growth of CSC cells. (a and b) Representative images and statistic results of CSC spheres with treatment of the miR-92a inhibitor and miR-92a + SOCS3 siRNA, **P < 0.01 vs CSC+NC group; #P < 0.05 vs CSC+miR-92a inhibitor. (c) qRT-PCR and (d) Western blot were utilized to estimate the relative expressions of CD133, SOX2, and OCT4 on the miR-92a inhibitor and miR-92a + SOCS3 siRNA treated SW480 and LoVo CSC cells. (f) Cell growth of different groups (Blank, CSC, CSC+inhibitor NC, CSC+miR-92a inhibitor, and CSC+miR-92a inhibitor+SOCS3 siRNA) were detected via EdU staining. (g) Western blot detection of SOCS3, AKT, STAT3, p-AKT, and p-STAT3 in different groups (Blank, CSC, CSC+inhibitor NC, CSC+miR-92a inhibitor, and CSC+miR-92a inhibitor+SOCS3 siRNA).
Discussion
In this study, miR-92a and SOCS3 were revealed to be upregulated and downregulated. In the functional analysis, miR-92a inhibition and SOCS3 overexpression were both demonstrated to attenuate the CRC CSC cell self-renewal capacity, and repressed CRC CSC cell proliferation. MiR-92a was revealed to bind and negatively modulate SOCS3 in CRC cells. Moreover, SOCS3 knockdown could abolish the suppressive impacts of the miR-92a inhibitor on the self-renewal capacity of CRC CSC cells. Hence, miR-92a facilitates the stemness of CRC cells by inhibiting SOCS3, suggesting that the miR-92a/SOCS3 axis might be a promising therapeutic target of CRC.
The existence of CSC cells and their profiles has been well documented by more and more literature referring to the initiation, progression, and recurrence of CRC [Citation24]. As reported, CSC cells are a subset of cancer cells, characterized by immature biomarkers, self-renewing, and strong drug-resistance [Citation25]. CSC cells can originate from non-stem, non-differentiated, or chemically treated tumor cells [Citation26]. The drug-resistance ability of cancer cells with significantly increased expression of stemness proteins, such as CD133, SOX2, and OCT4, is much stronger than those cells with either low or no stemness markers [Citation27,Citation28]. However, CRC stemness and its underlying regulatory mechanisms remains largely undetermined. miRNAs have important impacts on the stemness of various of human cancer cells [Citation29]. For example, miR-206 was reported to repress stemness and metastasis of breast tumor cells by affecting the MKL1/IL-11 cascade [Citation30]. Moreover, Guo JC et al. found that miR-448 can attenuate the self-renewing of HCC (hepatocellular carcinoma) stem cells by suppressing stemness maintenance, and the MAGEA6-mediated AMPK cascade was demonstrated to contribute to this phenomenon [Citation31]. Similarly, in CRC, miR-196a was reported to enhance the stemness of CRC cells by repressing ZG16 [Citation32]. MiR-92a was previously found to facilitate the tumorigenesis of CRC [Citation33,Citation34]. Nevertheless, whether miR-92a has an impact on CRC stemness remained uninvestigated. Here, we provided solid evidences that inhibition of miR-92a can destroy the stemness of CRC cells.
The JAK-STAT cascade was demonstrated to be a regulator of cytokine signaling [Citation35]. Inflammatory agents, including IL-1β, IL-6, and IFN-γ, play a key role in DNA methylation, and thereby have a close association with cancer, and the JAK-STAT cascade is a bridge between inflammation and cancer [Citation36]. Suppressor of cytokine signaling (SOCS) proteins could negatively regulate cytokine signaling mediated by the JAK-STAT cascade via a classical feedback loop [Citation21]. In particular, SOCS1 [Citation37,Citation38] and SOCS3 [Citation39], as potent inhibitors of JAKs, were reported to be played a critical role in various malignant processes, such as in cancers. SOCS1 suppressed metastatic progression of colorectal tumors by preventing the mesenchymal–epithelial transition (MET) [Citation40]. Moreover, Letellier et al [Citation41]. reported that SOCS2 and SOCS6 may be biomarkers in CRC. Knockdown of SOCS3 activates multiple oncogenes, resulting in the emergence of tumors [Citation42]. Low SOCS3 gene expression was reported to be associated with the metastasis of CRC cells [Citation43]. A study performed by Jablons DM et al. discovered that inhibition of SOCS3 by hypermethylation significantly facilitated the tumor growth of lung cancer [Citation44] and ulcerative colitis-related colorectal cancer [Citation45], suggesting that hypermethylation might be the predominate reason responsible for the downregulation of the SOCS3 protein, which further contributes to cancer progression by activating of the STAT3 cascade. Nevertheless, the mechanisms that modulate SOCS3, and the reason of the dysregulation of the JAK/STAT signaling cascade in CRC cells, remain undetermined. Recently, miR-196b-5p was found to facilitate stemness and resistance of CRC cells by targeting SOCS1 and SOCS3, leading to the activation of the STAT3 cascade [Citation23]. This finding suggested to us that the interaction between miRNAs and SOCS3 might be one of the main mechanisms underlying CRC stemness acquisition and maintenance. Here, we found that SOCS3 was dysregulated in CRC, and overexpression attenuated the CRC stemness. Moreover, SOCS3 can be bound by miR-92a, and knockdown of SOCS3 can abrogate the impacts of the miR-92a inhibitor on CRC stemness.
Conclusion
Our study suggested that miR-92a might have a critical role in the stemness of CRC cells by repressing SOCS3, improving our knowledge of CRC cell proliferation and stemness, and this result will contribute to developing effective therapeutic measures against CRC. As far as we know, this study is the first to report the exploration of the mechanism of miR-92a in the maintenance of stem-like property in colorectal cancer, which will provide more complete information for the involvement of miR-92a in the metastasis of colorectal cancer and lay a certain foundation for the pathogenesis of colorectal cancer.
Author Contributions
LFL and TZ designed the experiments. LFL performed most of the experiments. JXZ and HP performed the flow cytometry. LFL, HP and ZLL completed the informed consent and collected the clinical patients’ tissues. LFL, HPT and SLH collected the data, and XJX and QP analyzed the data. TZ confirmed the data analysis. LFL finished the draft. TZ revised the draft. All authors approved the version of manuscript before submission.
Conflicting Interest
The authors declare that they have no potential conflict of interest.
Acknowledgements
Thanks to Dr. Edward C. Mignot, Shandong University, for linguistic advice.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Brody H. Colorectal cancer. Nature. 2015;521(7551):S1.
- Roncucci L, Lmariani F. Prevention of colorectal cancer: how many tools do we have in our basket? Eur J Intern Med. 2015;26(10):752–756.
- Miller KD, Nogueira L, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin. 2019;69(5):363–385.
- Dawood S, Austin L, Lcristofanilli M. Cancer stem cells: implications for cancer therapy. Oncology (Williston Park). 2014;28(12):1101–1107.
- Vlashi E, Pajonk F. Cancer stem cells, cancer cell plasticity and radiation therapy. Semin Cancer Biol. 2015;31:28–35.
- Munro MJ, Wickremesekera SK, Peng L, et al. Cancer stem cells in colorectal cancer: a review. J Clin Pathol. 2018;71(2):110–116.
- Tutar Y. miRNA and cancer; computational and experimental approaches. Curr Pharm Biotechnol. 2014;15(5):429.
- Zhang Y, Yun Z, Gong L, et al. Comparison of miRNA evolution and function in plants and animals. Microrna. 2018;7(1):4–10.
- Wojciechowska A, Braniewska A, Akozar-Kaminska K. MicroRNA in cardiovascular biology and disease. Adv Clin Exp Med. (2017);26(5):865–874.
- Vishnoi A, Rani S. MiRNA biogenesis and regulation of diseases: an overview. Methods Mol Biol. 2017;1509:1–10.
- Zhu W, Long JL, Yin YT, et al. MicroRNA-34a suppresses the invasion and migration of colorectal cancer cells by enhancing EGR1 and inhibiting vimentin. Exp Ther Med. 2019;18(4):2459–2466.
- An HJ, Park M, Kim J, et al. miR5191 functions as a tumor suppressor by targeting RPS6KB1 in colorectal cancer. Int J Oncol. 2019. DOI:10.3892/ijo.2019.4865
- Wang LQ, Yu P, Li B, et al. miR-372 and miR-373 enhance the stemness of colorectal cancer cells by repressing differentiation signaling pathways. Mol Oncol. 2018;12(11):1949–1964.
- Li X, Guo S, Min L, et al. S.miR-92a-3p promotes the proliferation, migration and invasion of esophageal squamous cell cancer by regulating PTEN. Int J Mol Med. (2019);44(3):973–981.
- Sun B, Zhang J, Liu M, et al. Alkannin inhibits proliferation, migration and invasion of hepatocellular carcinoma cells via regulation of miR-92a. Biomed Pharmacother. 2019;114:108782.
- Sun L, Jin X, Xie L, et al. Swainsonine represses glioma cell proliferation, migration and invasion by reduction of miR-92a expression. BMC Cancer. 2019;19(1):247.
- Peng Q, Shen Y, Lin K, et al. Identification of microRNA-92a and the related combination biomarkers as promising substrates in predicting risk, recurrence and poor survival of colorectal cancer. J Cancer. 2019;10(14):3154–3171.
- Mahony R, Ahmed S, Diskin C, et al. SOCS3 revisited: a broad regulator of disease, now ready for therapeutic use? Cell Mol Life Sci. 2016;73(17):3323–3336.
- Yin Y, Liu W, Dai Y. SOCS3 and its role in associated diseases. Hum Immunol. 2015;76(10):775–780.
- Xu WP, D W. SOCS3: a potential therapeutic target for many human diseases. Yao Xue Xue Bao. 2011;46(7):747–752.
- Inagaki-Ohara K, Kondo T, Ito M, et al. SOCS, inflammation, and cancer. Jakstat. 2013;2(3):e24053.
- Chu Q, Shen D, He L, et al. Prognostic significance of SOCS3 and its biological function in colorectal cancer. Gene. 2017;627:114–122.
- Ren D, Lin B, Zhang X, et al. Maintenance of cancer stemness by miR-196b-5p contributes to chemoresistance of colorectal cancer cells via activating STAT3 signaling pathway. Oncotarget. 2017;8(30):49807–49823.
- Hu Y, Yan C, Mu L, et al. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS One. 2015;10(5):e0125625.
- De Angelis ML, Zeuner A, Policicchio E, et al. Cancer stem cell-based models of colorectal cancer reveal molecular determinants of therapy resistance. Stem Cells Transl Med. 2016;5(4):511–523.
- Li J, Condello S, Thomes-Pepin J, et al. Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell. 2017;20(3):303–314 e305.
- VAN Schaijik B, Davis PF, Wickremesekera AC, et al. Subcellular localisation of the stem cell markers OCT4, SOX2, NANOG, KLF4 and c-MYC in cancer: a review. J Clin Pathol. 2018;71(1):88–91.
- Amaya CN, Bryan BA. Enrichment of the embryonic stem cell reprogramming factors Oct4, Nanog, Myc, and Sox2 in benign and malignant vascular tumors. BMC Clin Pathol. 2015;15(1):18.
- Khan AQ, Ahmed EI, Elareer NR, et al. Role of miRNA-regulated cancer stem cells in the pathogenesis of human malignancies. Cells. 2019;8(8):840.
- Samaeekia R, Adorno-Cruz V, Bockhorn J, et al. miR-206 inhibits stemness and metastasis of breast cancer by targeting MKL1/IL11 pathway. Clin Cancer Res. 2017;23(4):1091–1103.
- Guo JC, Yang YJ, Zhang JQ, et al. microRNA-448 inhibits stemness maintenance and self-renewal of hepatocellular carcinoma stem cells through the MAGEA6-mediated AMPK signaling pathway. J Cell Physiol. 2019;234(12):23461–23474.
- Chen X, Du P, She J, et al. Loss of ZG16 is regulated by miR-196a and contributes to stemness and progression of colorectal cancer. Oncotarget. 2016;7(52):86695–86703.
- Chen E, Li Q, Wang H, et al. MiR-92a promotes tumorigenesis of colorectal cancer, a transcriptomic and functional based study. Biomed Pharmacother. 2018;106:1370–1377.
- Lv H, Zhang Z, Wang Y, et al. MicroRNA-92a promotes colorectal cancer cell growth and migration by inhibiting KLF4. Oncol Res. 2016;23(6):283–290.
- Huynh J, Etemadi N, Hollande F, et al. The JAK/STAT3 axis: a comprehensive drug target for solid malignancies. Semin Cancer Biol. 2017;45:13–22.
- Yu H, Lee H, Herrmann A, et al. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736–746.
- Hanada T, Kobayashi T, Chinen T, et al. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J Exp Med. 2006;203(6):1391–1397.
- Yoshida T, Ogata H, Kamio M, et al. SOCS1 is a suppressor of liver fibrosis and hepatitis-induced carcinogenesis. J Exp Med. 2004;199(12):1701–1707.
- Inagaki-Ohara K, Mayuzumi H, Kato S, et al. Enhancement of leptin receptor signaling by SOCS3 deficiency induces development of gastric tumors in mice. Oncogene. 2014;33(1):74–84.
- David M, Naudin C, Letourneur M, et al. Suppressor of cytokine signaling 1 modulates invasion and metastatic potential of colorectal cancer cells. Mol Oncol. 2014;8(5):942–955.
- Letellier E, Schmitz M, Baig K, et al. Identification of SOCS2 and SOCS6 as biomarkers in human colorectal cancer. Br J Cancer. 2014;111(4):726–735.
- Sutherland KD, Vaillant F, Alexander WS, et al. c-myc as a mediator of accelerated apoptosis and involution in mammary glands lacking Socs3. EMBO J. 2006;25(24):5805–5815.
- Zhang L, Li J, Li L, et al. IL-23 selectively promotes the metastasis of colorectal carcinoma cells with impaired Socs3 expression via the STAT5 pathway. Carcinogenesis. 2014;35(6):1330–1340.
- He B, You L, Uematsu K, et al. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc Natl Acad Sci U S A. 2003;100(24):14133–14138.
- Li Y, Deuring J, Peppelenbosch MP, et al. IL-6-induced DNMT1 activity mediates SOCS3 promoter hypermethylation in ulcerative colitis-related colorectal cancer. Carcinogenesis. 2012;33(10):1889–1896.