
ABSTRACT
The COVID-19 new variants spread rapidly all over the world, and until now scientists strive to find virus-specific antivirals for its treatment. The main protease of SARS-CoV-2 (Mpro) exhibits high structural and sequence homology to main protease of SARS-CoV (93.23% sequence identity), and their sequence alignment indicated 12 mutated/variant residues. The sequence alignment of SARS-CoV-2 main protease led to identification of only one mutated/variant residue with no significant role in its enzymatic process. Therefore, Mpro was considered as a high-profile drug target in anti-SARS-CoV-2 drug discovery. Apigenin analogues to COVID-19 main protease binding were evaluated. The detailed interactions between the analogues of Apigenin and SARS-CoV-2 Mpro inhibitors were determined as hydrogen bonds, electronic bonds and hydrophobic interactions. The binding energies obtained from the molecular docking of Mpro with Boceprevir, Apigenin, Apigenin 7-glucoside-4’-p-coumarate, Apigenin 7-glucoside-4’-trans-caffeate and Apigenin 7-O-beta-d-glucoside (Cosmosiin) were found to be −6.6, −7.2, −8.8, −8.7 and −8.0 kcal/mol, respectively. Pharmacokinetic parameters and toxicological characteristics obtained by computational techniques and Virtual ADME studies of the Apigenin analogues confirmed that the Apigenin 7-glucoside-4’-p-coumarate is the best candidate for SARS-CoV-2 Mpro inhibition.
1. Introduction
Coronavirus infection 2019 (COVID-19) has caused more than 237,383,711 confirmed cases and 4,842,716 deaths until the 10 October 2021 in the world (https://covid19.who.int/). Therapies against coronavirus can be classified into two strategies such as drugs acting on the immune system and drugs targeting the virus. Vaccine development has been accelerated, with more than 6,364,021792 vaccine candidates (https://covid19.who.int/). Unfortunately, despite the high level of vaccination and the reduction of the transmission, these therapies may lose their efficiency if the virus mutates and/or changes its antigenicity as observed with the South African variant (variant B.1.351) [Citation1], ‘Epsilon’ variant (B.1.429) in Taiwan [Citation2] and ‘Mu’ variant (B.1.621) in Colombia [Citation3]. The key SARS-CoV-2 targets for therapies comprise a structural protein (responsible for replication, transcription and host cell recognition) and three nonstructural proteins (RdRp, PLpro and 3 CLpro) [Citation4]. It was recently found that the main protease of this virus (Mpro) plays a crucial role in SARS-CoV gene expression and replication [Citation5]. Moreover, genome sequence analyses revealed that COVID-19 shares a high level of sequence similarities with SARS-CoV and MERS-CoV [Citation6]. Mpro has been validated as an attractive target for anti-SARS-CoV drug design, and a variety of inhibitors have been developed [Citation7,Citation8], especially considering the re-use of existing MERS and SARS Mpro inhibitors. Recently, approved inhibitors including Darunavir [Citation9], Danoprevir [Citation10] and Boceprevir [Citation11] have been used to treat COVID-19 patients. Boceprevir is the recommended treatment as inhibitor for the Mpro SARS-CoV-2. However, natural sources such as micro-organisms [Citation12], algae [Citation13] and plants [Citation14] need to be explored to produce new pharmaceutical treatments against SARS-CoV-2. Natural active constituents played a crucial role in drug discovery to treat diverse diseases because of their natural characteristics, lower toxicity and fewer drug remnants in body [Citation15]. Several natural molecules were reported to be able of inhibiting the main protease of Sars-CoV-2 such as quercetin, gallocatechin gallate and epigallocatechin gallate with IC50 values of 73 µM, 47 µM and 73 µM, respectively [Citation16,Citation17]. The Apigenin (4′,5,7-trihydroxyflavone), a glycoside from the flavones class, is found in many fruits and vegetables, more particularly in Tunisian plants such as Retama raetam Forssk [Citation18], Zizyphus lotus L. [Citation19] and in seven principal Tunisian olive varieties [Citation20]. The Apigenin produced from plants have antioxidant [Citation21], anticancer [Citation22–24], anti-inflammatory [Citation25] and anti-hyperglycemic activities [Citation26] and were previously used as compounds in drug discovery. Recent studies have also shown antiviral efficacy of Apigenin against several viruses [Citation26–32]. In addition, Khandelwal et al. (2020) [Citation33] described that Apigenin have antiviral effects against buffalopox virus.
In-silico approaches have accelerated the process of drugs finding compared to the conventional methods [Citation34]. Molecular docking has been used to predict the binding models of inhibitors to several targets. They have been successfully used to design or to study the interaction between Mpro and promising inhibitors [Citation7,Citation35]. This study was undertaken to investigate the viral adaptation especially the genetic stability of SARS-CoV-2 main proteases from different variants as well as the inhibitory activities of Apigenin and its analogues against this target via in-silico studies (binding energies, detailed interactions and pharmacokinetic properties).
2. Materials and methods
2.1. Amino acid sequence analyses and homology modeling
Sequence analysis (GenBank 6WTT-A [Citation20], QVD51579.1, QWF00346.1, QMV29895.1, QRX05355.1, QTN92506.1, QRW91276.1, QNN90050.1, QNN90062.1 and QNN90074.1) and multiple alignments were performed using the BLAST and CLUSTALW programs [Citation36]. The prediction of the protein secondary structure was performed using the DSSP program [Citation37], while the editing of the alignment including the superimposition of secondary structures was conducted using the ESPript 3.0 program [Citation38]. The automated comparative protein structure homology modeling server, Geno3D (https://geno3d-prabi.ibcp.fr) generated the 3D structure models of SARS-CoV-2 Mpro using the published structure as template (PDB-code 6WTT) [Citation39]. PyMOL (http://www.pymol.org) and ViewerLite 5.0 softwares (https://www.3dsbiovia.com) were used to visualize and analyze the generated model structures and to construct the graphical presentations and illustrative figures.
2.2. Docking methodology
The three-dimensional x-ray crystal structure of SARS-CoV-2 Mpro (pdb code: 6WTT) was retrieved in pdb format from Protein Data Bank with resolution 2.15 Å [Citation39]. After that, the co-crystallized ligand of the SARS-CoV-2 Mpro structure was extracted. Then, it was prepared in AutodockVina by removal of water and solvent molecules, addition of polar hydrogens, removal of the bound ligand and partial charge assignment and saved as.pdbqt format using AutodockVina to be included as a reference in the virtual screening. The grid box was defined by selecting the co-crystallized inhibitors to keep the center of each docked Apigenin analogues with same dimensions of binding box. Moreover, the grid box center was adjusted X = 4. 9, Y = 27.64 and Z = −11.206 with dimensions for SARS-CoV-2 Mpro. Its size was set to 60 × 50 x 50 Angstroms to cover the active site. The structure of Apigenin and Apigenin analogues were downloaded from the PubChem search (https://pubchem.ncbi.nlm.nih.gov/). AutodockVina program was performed between Apigenin analogues and SARS-CoV-2 Mpro for molecular docking analysis such as binding types of interactions, binding energies, inhibition activities, ligand efficiency and distances. Molecular docking scores were set as AutoDock tools of the molecular graphics laboratory software package by keeping the analogue flexible [Citation40]. Boceprevir was used as control to compare the molecular docking results with the Apigenin analogues.
2.3. LigPlot analysis
Academic licensed LigPlot software was obtained from https://www.ebi.ac.uk/. This program is used to provide 2-D representation of protein–ligand interactions, intermolecular interactions like hydrogen bonding, hydrophobic interactions and atom accessibilities of their strengths [Citation41].
2.4. In-silico Osiris/Molinspiration and ADMET analysis
Osiris and Molinspiration analyses are performed to describe 2D models and to indicate the type of pharmacophore site [Citation42,Citation43]. These analyses are employed to predict pharmacophore site and biological activity of the apigenin analogues and to determine the drug-likeness score. The acute toxicity in rodent models and chemical classification of the test compounds were predicted by GUSAR [Citation44]. It analyzes compounds based on the quantitative neighborhoods of atom descriptors and prediction of activity spectra for substance algorithm and correlates the obtained results with the SYMYX MDL toxicity database. Furthermore, it classifies them based on the Organization for economic co-operation and development (OECD) chemical classification manual. The pharmacokinetic properties of the apigenin analogues were achieved with using the SwissADME, which is an open online tool (http://www.swissadme.ch). The ADME properties define blood–brain barrier (BBB) permeability and passive human gastrointestinal absorption (HIA) as well as substrate or nonsubstrate permeability glycoprotein (P-gp) and cytochrome P450 (CYP) [Citation42].
3. Results
‘This study aims to investigate the virus mutations and/or antigenicity changes and find the conserved targets. According to molecular and modeling studies, we confirm the genetic stability of SARS-CoV-2. Main proteases from different variant and that it constitutes a high-profile drug target. For faster and more cost-efficient drug discovery, we used the in-silico approach for prediction of the inhibitory activities of apigenin and its analogues against this target. The study confirms the potential of the apigenin 7-glucoside-4’-p-coumarate to inhibit Mpro SARS-CoV-2. It was observed that this analogue obtained good pharmacokinetic and toxicological characteristics. These finding suggest the ability to substitute boceprevir by this natural product present in several local plants for SARS-CoV-2 treatment.’
3.1. Conserved sequence among Mpro SARS-CoV-2
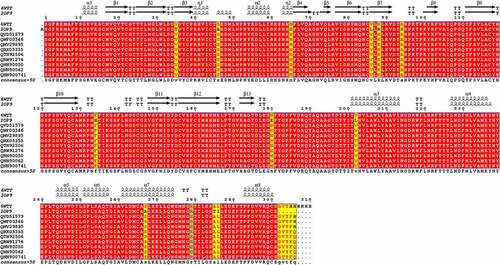
A thorough comparison of the primary and secondary structures of the 3 CL protease sequence (Mpro) was carried out (). The alignment of 10 sequences of Mpro from different variant of SRARS-Cov-2 and the sequence of Mpro from SARS-CoV (2OP9 [Citation45]) showed the presence of 12 mutations (Identity = 93.23%). However, all SARS-CoV-2 Mpro sequences are 100% conserved except for two Tunisian variants showing only 1 residue of 306 different from that of SARS-CoV-2 (identity = 97.74%) ().
Figure 1. Structure-based multiple sequence alignment of SARS-CoV-2 Mpro from different variant (6WTT, 2OP9, QVD51579.1, QWF00346.1, QMV29895.1, QRX05355.1, QTN92506.1, QRW91276.1, QNN90050.1, QNN90062.1 and QNN90074.1). Residues invariable among sequences are typed in white on a red background; residues conserved within each group are typed in red on a yellow background. The residue mutated in this study (R279) is indicated in light blue. Secondary structure elements from of Mpro structure are indicated at the top of the alignment with SARS-CoV-2 main proteases (PDB code: 6WTT) .
3.2. Structural aspects
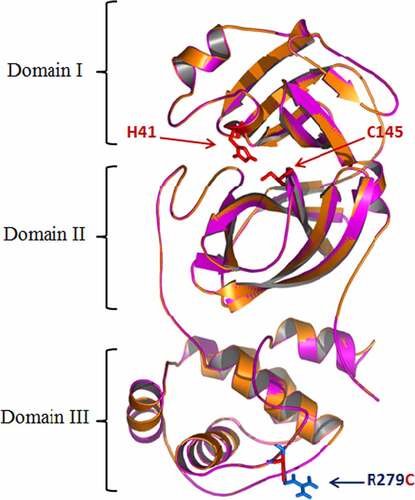
The modeling of SARS-CoV-2 Main proteases from different variants and the characterization of the mutations structural impacts was investigated. Mpro model (306 residues) was built by the aid of the automated homology modeling, Geno3D, web server using SARS-CoV-2 Main protease (PDB ID: 6WTT, chain A) as homolog. The Mpro sequence exhibited high identity with that of the template (99.76%) suggesting the high-quality models that could be obtained. The obtained model showed a perfect superimposition of the Cα with 6WTT regarding to the very low RMSD (root mean square deviation) value estimated at 0.914 Å. The Main protease of SARS-CoV-2 was showed as composed of three domains (). The domains I and II have an antiparallel β-barrel fold. The cleft between these domains generates the substrate-binding site. Domain III has a globular structure formed by five α-helices. A loop (residues 183–198) connected this domain to domain II. Mpro SARS-CoV-2 has a C145-H41 catalytic dyad. Modeling results showed that the mutation (R279C) is situated in domain III, far of the active site (). These data confirmed that the structure is similar to main protease of SARS-CoV-2 [Citation46] and the genetic stability of main proteases of SARS-CoV-2 from different variants.
Figure 2. Superimposition of structure of the SARS-CoV-2 Mpro (pdb: 6WTT) (Orange) and model of the SARS-CoV-2 3 Mpro fromTunisian variant QNN90062.1 (pink). The catalytic dyad (Cys-145 and His-41) is colored in red.
3.3. Molecular docking studies
In this study, the four Apigenin analogues along with the Boceprevir were investigated as potential inhibitors of the Mpro SARS-CoV-2 using Autodock Vina tools. Boceprevir was showed, using enzyme inhibition and co-crystal structure analyses, to inhibit replication of SARS-CoV-2 in cell culture [Citation11,Citation47]. In this study, all Apigenin analogues as well as Boceprevir were docked using similar optimized docking conditions. All the docked poses into the binding site of Mpro SARS-CoV-2 were analyzed with identified docking search algorithms and scoring functions ( and ).
Table 1. Details of the Apigenin analogues compounds and Boceprevir from the docking analysis with Mpro substrate binding site
Figure 3. Interactions of the SARS-CoV-2 Mpro (pdb: 6WTT) with Boceprevir (a), Apigenin (b), Apigenin 7-glucoside-4’-p-coumarate (c), Apigenin 7-glucoside-4’-trans-caffeate (d) and Apigenin 7-O-beta-D-glucoside (Cosmosiin) (e) .
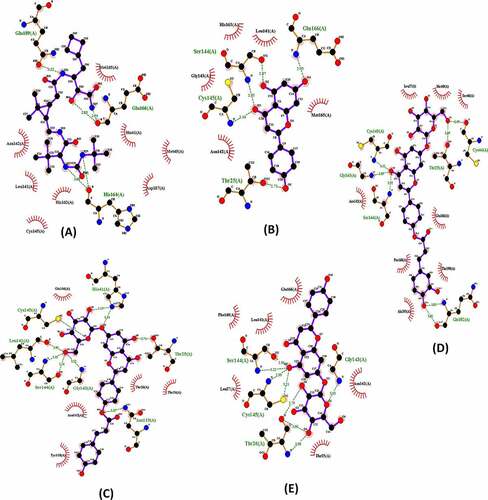
The poses made using Ligplot are shown in for Boceprevir and the four ligands. Concerning Boceprevir, the best pose with the lowest binding energy (−6.6 Kcal/mol) showed that the hydroxyl and amide groups, resulting from the covalent addition to the α-ketoamide, form two hydrogen bonds with the main chain of Glu166. The tert-butyl group is relatively solvent exposed and forms two hydrogen bonds with His164. The amide bond on the main chain of Boceprevir forms hydrogen bond with the side chain of Gln189. Many other residues forming hydrophobic interactions (like His41, Met49, Met165, Asp163) stabilize the conformation of the ligand. Apart from our docked models, other published Mpro Docked results with boceprevir could be found [Citation11,Citation39]. These complex structures show highly similar binding poses to ours. However, structures containing Boceprevir are now available [Citation47,Citation48]. These complex structures show that the carbonyl of the electrophilic α-ketoamide could form a covalent bond with the sulfur of the catalytic residue Cys145 stabilizing the structure. The oxygen of the same group forms two hydrogen bonds with the main chain amides of Cys145 and Gly143. Hence, the oxyanion hole with its S1, S1’ and S2 pockets is occupied. Nevertheless, and as found by previous studies, the cyclobutylmethyl group of boceprevir is inserted superficially into the S1 pocket and is relatively solvent exposed ().
Figure 4. The active site of SARS-CoV-2 (a) bound with Boceprevir (b), Apigenin (c), Apigenin 7-glucoside-4’-p-coumarate (d), Apigenin 7-O-beta-D-glucoside (Cosmosiin) (e) and Apigenin 7-glucoside-4’-trans-caffeate (f) .
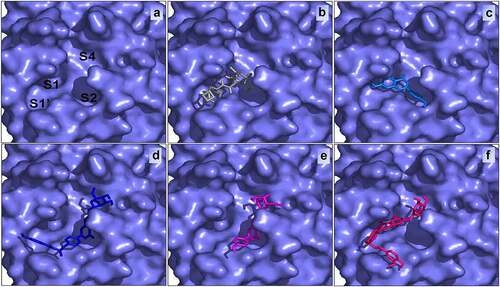
According to , the best binding energy analogue was the Apigenin 7-glucoside-4’-p-coumarate (−8.8 kcal/mol) followed by the Apigenin 7-glucoside-4’-trans-caffeate (−8.7 kcal/mol). These two Apigenin analogues displayed with residues in active site of Mpro SARS-CoV-2, respectively, 9 and 7 hydrogen bonds.
For the Apigenin 7-glucoside-4’-trans-caffeate, among eight hydrophobic interactions, an amide π-stacked interaction (with Leu167) and a π-alkyl interaction (with Pro168) were established with the dihydroxyphenyl moiety of the caffeate. In addition, a π–π Tshaped was detected with His 41 established with the benzopyran ring of the Apigenin moiety. For the Apigenin 7-glucoside-4’-p-coumarate, five residues were found to be involved in hydrophobic interactions, and a π–π stacked interaction was detected between Tyr118 and the hydroxyphenyl ring of the coumarate moiety. Finally, concerning Cosmosiin (Apigenin 7-O-beta-d-glucoside), among other hydrophobic contacts, a π-sigma bond was established between Asn142 and benzopyran ring of the Apigenin. In all cases, the sugar moieties were involved essentially in hydrogen bonds.
3.4. Pharmacokinetic studies
The pharmacokinetic and toxicity properties of the Apigenin analogues with best binding energies were evaluated as potential drug candidates. General Unrestricted Structure–Activity Relationships (GUSAR) software [Citation44] was used for quantitative in-silico toxicity prediction for Boceprevir, Apigenin and Apigenin analogues in rats with four types of administration (intraperitoneal, intravenous, oral and subcutaneous). As displayed in , the different LD50 values suggests that availability of the tested inhibitors for metabolism by the liver is a major factor for its toxicity. The LD50 value of Apigenin 7-glucoside-4’-p-coumarate and Cosmosiin was higher than Boceprevir for intravenous (IV), oral, and subcutaneous (SC) routes of administration. Only the LD50 value of Cosmosiin was higher through intraperitoneal (IP) route. By the OECD chemical classification system, only the Apigenin 7-glucoside-4’-trans- are class 4 when administered through Intraperitoneal compared to class 5 for the other compounds. The toxicity profile of these compounds is relatively low, and they require high doses to elicit toxic responses.
Table 2. Prediction of acute toxicity in-silico by GUSAR in rodent models and chemical classification of compounds
The pharmacophore features and drug-like properties of the Apigenin and its analogues were performed with Molinspiration and Osiris Property Explorer [Citation49]. The cLogP values (which is octanol/water partition coefficient) of the Apigenin analogues were found lower than 5.0 (). This finding suggests that these analogues have rational high absorption and permeability [Citation50,Citation51].
Table 3. Drug likeliness properties of the Apigenin analogues by swissAdme, osiris and Molinspiration
Solubility is known to be a significant parameter for drug design and pharmacology due to the potential absorption and distribution characteristics. Thus, soluble drugs are preferred in drug manufacturing [Citation52]. The solubility values of most drugs sold in the market are greater than −4.0 and the solubility values of Apigenin and Cosmossin were −2.86 and −2.74, respectively. The solubility of Apigenin 7-glucoside-4’-p-coumarate and Apigenin 7-glucoside-4’-trans-caffeate values was also close to −4 ().
Furthermore, Ion Channel Modulator, Human G-protein coupled receptors (GPCRs) ligands, Nuclear Receptor Ligand, Kinase Inhibitor, Protease Inhibitor and Enzyme inhibitors of the Apigenin analogues were illustrated with the prediction bioactivity scores using online-site Molinspiration (). As presented in , metabolic enzymes such as Cytochrome P450 (CYP) and the transporter class P-glycoprotein (P-gp) were equally assessed in this study. Boceprevir, Apigenin 7-glucoside-4’-p-coumarate, Apigenin 7-glucoside-4’-trans-caffeate and Cosmossin were not found to be inhibitors of CYP except CYP2C9 inhibitor confirming the goodness of their transport in the intestine [Citation53].
4. Discussion
The virus mutates and/or changes its antigenicity causing loss of the vaccination efficiency. Many experimental and computational efforts done to identify a genetic stable target for anti-SARS-CoV drug design were provided. In this work, the genetic stability of Main proteases of SARS-CoV-2 from different variants was confirmed. In addition, the superimposition of the crystal structures of Mpro with and without ligands has RMSD ranging from 0.26 to 0.38 Å, indicated that the binding pocket is pre-shaped [Citation54]. Thus, Mpro is an attractive target for anti-SARS-CoV drug design. The inhibitors of this enzyme can be found by structure-based design [Citation35], by enzymatic assay of existing inhibitors of other virus main protease such as HIV or HVC protease inhibitors [Citation55] or by screening of chemical database using docking approaches. Boceprevir is generally used as reference molecule especially for its low renal and hepatic toxicity [Citation56]. The recent reports suggest that boceprevir inhibits the enzymatic activity of Mpro with an IC50 value of 4.13 μM [Citation55]. This result was confirmed by co-crystallization of Mpro and Boceprevir [Citation57].
In order to find natural molecules as an alternative of Boceprevir, the Apigenin and Apiginin analogues were investigated as potential inhibitors of the Mpro SARS-CoV-2. The docking results showed that the binding energy values (−8.8 to −7.2 kcal/mol) are better than that of Boceprevir and two Boceprevir analogues (PubChem ID 57841991 and 58,606,278) with binding energies of −6.6, −7.2 and −7.5 kcal/mol, respectively [Citation11].
Apigenin 7-glucoside-4’-p-coumarate occupies the S1′, S2, and S4 subsites while Boceprevir filled the S1, S1′, and S2 subsites of SARS-CoV-2 Mpro active site. Generally, small size inhibitors may bind only at S1 and S2 subsites [Citation11,Citation58]. Boceprevir with its medium size may bind to S1′, S1, and S2 subsites [Citation59,Citation60], while some large inhibitors may bind to S1’, S1, S2, and may extend through S3 subsites as for example alpha-ketoamide (PDB: 6Y2F) [Citation61,Citation62]. These compounds have dissimilar structures and sizes, yet they bind and inhibit the activity of SARS-CoV-2 Mpro.
All hydrogen bonds and hydrophobic interactions are shown in . The common residues in active site of Mpro SARS-CoV-2 stabilizing all analogues best poses are His 41, Ser144, Gly143, Thr25, Thr26, Glu166, Asn142 and especially the catalytic Cys145. The latter residue, because of its proximity to the ligand, was also suspected to establish covalent bond with certain atoms thanks to its thiol group as it has been demonstrated in the crystallization experiments but not been detected by docking experiments [Citation32]. All the aforementioned residues are identified as key interactions between SARS-CoV-2 main protease and inhibitor drug candidates [Citation34].
We observed for all Apigenin analogues strong hydrogen bonding. In addition, although the thiol group of Cys145 which was found to interact via hydrogen bonds, it is suspected to be able for covalent bonds as confirmed by previous structural studies. Gly143, Ser144, Glu166, Asn142 and Cys145 also interact with each inhibitor and are probably implicated in its stabilization. The best binding energy molecule such as Apigenin 7-glucoside-4’-p-coumarate occupies the S1′, S2, and S4 subsites ().
Apigenin 7-glucoside-4’-p-coumarate was found to have the best desirable pharmacokinetic properties such as low hepatotoxicity, good aqueous solubility, high intestinal absorption, non CYP2D6 binding and inability to cross the BBB besides good binding properties with SARS-CoV-2 Mpro active site. Therefore, we demonstrate that the Apigenin 7-glucoside-4’-p-coumarate identified by our in-silico study have potential against COVID-19 and may bind and inhibit the SARS-CoV-2 main protease.
Similar studies on structure–activity relationship analysis demonstrated that polyphenols and flavonoid (naringenin, rutin and tangeretin) from Citrus and Curcuma spp. have high affinity to the active site of the Mpro [Citation63].
Natural plant medicines are shown to ameliorate the recovery of infected person and to prevent SARS-CoV-2 infection of healthy persons as well as to improve the health state of patients with mild or severe symptoms [Citation64]. Among others, plant originated cinnamic amides, flavonoids, chalcones, tanshinones and diarylheptanoids are shown to inhibit the PLpro one of the nonstructural proteins encoded by the SARS-CoV-2 genome [Citation65]. However, other natural products are demonstrated to inhibit the 3 CL(pro), another nonstructural protein of the SARS-CoV-2. As examples alkylated Chalcones, phlorotannins and bioflavonoids were described in the literature [Citation66]. Most of the studies were performed using in-silico approaches and few of them combined in vitro and/or in vivo experiments. Most of the studies were conducted with isolated natural compounds and phenolic compounds were the most frequently reported. In this line, our study confirmed the potential of the Apigenin 7-glucoside-4’-p-coumarate to inhibit Mpro SARS-CoV-2. It was observed that this analogue obtained good pharmacokinetic and toxicological characteristics. These finding suggest the ability to substitute Boceprevir by this natural product present in several local plants for SARS-CoV-2 treatment. However, the in-silico study evaluating the anti-SARS-CoV-2 effect is still insufficient even if this natural products could be considered as promising anti-SARS-CoV-2 agents.
5. Conclusion
Our study confirmed the potential of the Apigenin 7-glucoside-4’-p-coumarate, naturally present in different Tunisian plants, to inhibit Mpro SARS-CoV-2 with the best binding energy. It was observed that this analogue obtained good results in terms of its toxicity properties. These finding suggest the ability to substitute Boceprevir by apigenin 7-glucoside-4’-p-coumarate for SARS-CoV-2 treatment.
Highlights
Main protease of SARS-CoV-2 (Mpro) is a high-profile drug target.
Good pharmacokinetic and toxicological characteristics of Apigenin analogues.
Apigenin 7-glucoside-4’-p-coumarate may be a good candidate for Mpro inhibition.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Hoffmann M, Arora P, Groß R, et al. SARS-CoV-2 variants B. 1.351 and P. 1 escape from neutralizing antibodies. Cell. 2021;184(9):2384–2393. e12.
- Akhmetzhanov AR, Jung S-M, Cheng H-Y, et al. A hospital-related outbreak of SARS-CoV-2 associated with variant Epsilon (B. 1.429) in Taiwan: transmission potential and outbreak containment under intensified contact tracing, January–February 2021. Inter J Infect Dis. 2021;110:15–20.
- Laiton-donato K, Franco-munoz C, Alvarez-diaz DA, et al. Characterization of the emerging B. 1.621 variant of interest of SARS-CoV-2. Infect Genet Evol. 2021;95:105038 .
- Andersen KG, Rambaut A, Lipkin WI, et al. The proximal origin of SARS-CoV-2. Nat Med. 2020;26(4):450–452.
- Yuan M, Hejun LIU, WU NC, et al. Structural basis of a shared antibody response to SARS-CoV-2. Science. 2020;369(6507):1119–1123.
- Aiping WU, Peng Y, Huang B, et al. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe. 2020;27(3):325–328.
- Hongbo LIU, Fei YE, Qi SUN, et al. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. J Enzyme Inhib Med Chem. 2021;36(1):497–503.
- Xueting YAO, Fei YE, Zhang M, et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin Infect Dis. 2020;71(15):732–739.
- Meyer DE, Sandra BOJKOVA, Denisa CINATL, et al. Lack of antiviral activity of darunavir against SARS-CoV-2. Inter J Infect Dis. 2020;97:7–10.
- Chen H, Zhang Z, Wang L, et al. First clinical study using HCV protease inhibitor danoprevir to treat COVID-19 patients. Medicine (Baltimore). 2020;99(48):e23357.
- Borkotoky S, Banerjee M, Modi GP, et al. Identification of high affinity and low molecular alternatives of boceprevir against SARS-CoV-2 main protease: a virtual screening approach. Chem Phys Lett. 2021;770:138446.
- Daverey A, Dutta K, Joshi S, et al. Sophorolipid: a glycolipid biosurfactant as a potential therapeutic agent against COVID-19. Bioengineered. 2021;12:9550–9560.
- Chia WY, Hanz KOK, Chew KW, et al. Can algae contribute to the war with Covid-19? Bioengineered. 2021;12(1):1226–1237.
- Tianyu Z, Liying G, . Identifying the molecular targets and mechanisms of xuebijing injection for the treatment of COVID-19 via network pharmacology and molecular docking. Bioengineered. 2021;12(1):2274–2287.
- Ben Halima N, Ben Slima A, Moalla I, et al. Protective effects of oat oil on deltamethrin-induced reprotoxicity in male mice. Food Function. 2014;5:2070–2077.
- Fendri I, Chamkha M, Bouaziz M, et al. Olive fermentation brine: biotechnological potentialities and valorization. Environ Technol. 2013;34:181–193.
- Ghosh R, Chakraborty A, Biswas A, et al. Evaluation of green tea polyphenols as novel Corona virus (SARS CoV-2) main protease (Mpro) inhibitors–an in silico docking and molecular dynamics simulation study. J Biomol Struct Dyn. 2020;12:4362–4374.
- Kassem M, Mosharrafa SA, Saleh NAM, et al. Two new flavonoids from Retama raetam. Fitoterapia. 2000;71(6):649–654.
- Najjaa H, Abdelkarim BA, Doria E, et al. Phenolic composition of some Tunisian medicinal plants associated with anti-proliferative effect on human breast cancer MCF-7 cells. EuroBiotech. J. 2020;4:104–112.
- Abaza L, Talorete TPN, Yamada P, et al. Induction of growth inhibition and differentiation of human leukemia HL-60 cells by a Tunisian gerboui olive leaf extract. Biosci Biotechnol Biochem. 2007;71(5):1306–1312.
- Fidelis QC, Faraone I, Russo D, et al. Chemical and biological insights of Ouratea hexasperma (A. St.-Hil.) Baill.: a source of bioactive compounds with multifunctional properties. Nat Prod Res. 2019;33(10):1500–1503.
- Imran M, Gondal ASLAM, Tanweer ATIF, et al. Apigenin as an anticancer agent. Phytother Res. 2020;34(8):1812–1828.
- Salehi B, Venditti A, Sharifi-rad M, et al. The therapeutic potential of apigenin. Int J Mol Sci. 2019;20(6):1305.
- Singh D, Khan MA, Et Siddique HR. Apigenin, a plant flavone playing Noble roles in Cancer prevention via modulation of key cell signaling networks. Recent Pat Anticancer Drug Discov. 2019;14(4):298–311.
- Ratana LIM, Barker G, Wall CA, et al. Dietary phytophenols curcumin, naringenin and apigenin reduce infection-induced inflammatory and contractile pathways in human placenta, foetal membranes and myometrium. Mol Hum Reprod. 2013;19(7):451–462.
- Villa-rodriguez JA, Kerimi A, Abranko L, et al. Acute metabolic actions of the major polyphenols in chamomile: an in vitro mechanistic study on their potential to attenuate postprandial hyperglycaemia. Sci Rep. 2018;8(1):1–14.
- Wenwen DAI, Jinpeng BI, Fang LI, et al. Antiviral efficacy of flavonoids against enterovirus 71 infection in vitro and in newborn mice. Viruses. 2019;11(7):625.
- Qian S, Wenchun FAN, Qian P, et al. Apigenin restricts FMDV infection and inhibits viral IRES driven translational activity. Viruses. 2015;7(4):1613–1626.
- Zhang W, Qiao H, Yuanzi LV, et al. Apigenin inhibits enterovirus-71 infection by disrupting viral RNA association with trans-acting factors. PloS One. 2014;9(10):e110429.
- Chung-Chun WU, Fang C-Y, Cheng Y-J, et al. Inhibition of Epstein-Barr virus reactivation by the flavonoid apigenin. J Biomed Sci. 2017;24(1):1–13.
- Shibata C, Ohno M, Otsuka M, et al. The flavonoid apigenin inhibits hepatitis C virus replication by decreasing mature microRNA122 levels. Virology. 2014;462:42–48.
- Hakobyan A, Arabyan E, Avetisyan A, et al. Apigenin inhibits African swine fever virus infection in vitro. Arch Virol. 2016;161(12):3445–3453.
- Khandelwal N, Chander Y, Kumar R, et al. Antiviral activity of Apigenin against buffalopox: novel mechanistic insights and drug-resistance considerations. Antiviral Res. 2020;181:104870.
- Le Minh BUI, Phung THITHU, Huong HOTHI, et al. Recent findings and applications of biomedical engineering for COVID-19 diagnosis: a critical review. Bioengineered. 2021;12(1):8594–8613.
- Wenhao DAI, Zhang B, Jiang X-M, et al. Structure-based design, synthesis and biological evaluation of peptidomimetic aldehydes as a novel series of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368(6497): 1331–1335.
- Thompson JD, Higgins DG, Et Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680.
- Kabsch WESANDER, Christian. Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers. 1983;22(12):2577–2637.
- Robert XEGOUET, Patrice. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42(W1):W320–W324.
- Chunlong MA, Sacco MD, Hurst B, et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30(8):678–692.
- Seeliger DEDEGROOT, Bert L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 2010;24(5):417–422.
- Wallace AC, Laskowski RA, Et Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng Des Sel. 1995;8(2):127–134.
- Daina A, Michielin O, Et Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1):1–13.
- Sander T, Freyss J, Von Korff M, et al. OSIRIS, an entirely in-house developed drug discovery informatics system. J Chem Inf Model. 2009;49(2):232–246.
- Lagunin A, Zakharov A, Filimonov D, et al. QSAR modelling of rat acute toxicity on the basis of PASS prediction. Mol Inform. 2011;30(2‐3):241–250.
- Goetz DH, Choe Y, Hansell E, et al. Substrate specificity profiling and identification of a new class of inhibitor for the major protease of the SARS coronavirus. Biochemistry. 2007;46(30):8744–8752.
- Zhenming JIN, Xiaoyu DU, XU Y, et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293.
- Oerlemans R, Ruiz-moreno AJ, Cong Y, et al. Repurposing the HCV NS3–4A protease drug boceprevir as COVID-19 therapeutics. RSC med chem. 2021;12(3):370–379.
- Kneller DW, Galanie S, Phillips G, et al. Malleability of the SARS-CoV-2 3CL Mpro active-site cavity facilitates binding of clinical antivirals. Structure. 2020;28(12):1313–1320. e3.
- Uysal UD, Ercengiz D, Karaosmanoğlu O, et al. Theoretical and experimental electronic transition behaviour study of 2-((4-(dimethylamino) benzylidene) amino)-4-methylphenol and its cytotoxicity. J Mol Struct. 2021;1227:129370.
- Erickson HK, Lambert JM. ADME of antibody–maytansinoid conjugates. The AAPS journal. 2012;14(4):799–805.
- Borah P, Hazarika S, Deka S. Application of Advanced Technologies in Natural Product Research: A Review with Special Emphasis on ADMET Profiling. Curr Drug Metab. 2020;21(10):751–767.
- Coltescu A-R, Butnariu M, Et Sarac I. The importance of solubility for new drug Molecules. Biomed Pharmacol J. 2020;13(2):577–583.
- Zhenming KY, Xiaoyu D, Yechun X, et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. 2020;582:289–293.
- Arya R, Kumari S, Pandey B, et al. Structural insights into SARS- CoV-2 proteins. J Mol Biol. 2021;433(2):166725.
- Chunlong MA, Sacco MD, Hurst B, et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30(8):678–692.
- Rizza SA, Talwani R, Nehra V, et al. Boceprevir. Drugs Today (Barc). 2011;47(10):743–751.
- Patil R, Chikhale P. Computational and network pharmacology analysis of bioflavonoids as possible natural antiviral compounds in COVID-19. Inform Med Unlocked. 2021; 22: 100504.
- Zhenming JIN, Zhao Y, Yuan SUN, et al. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat Struct Mol Biol. 2020;27(6):529–532.
- Rathnayake AD, Zheng J, Yunjeong KIM, et al. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV–infected mice. Sci Transl Med. 2020;12(557):1–11.
- Vuong W, Khan MB, Fischer C, et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat Commun. 2020;11(1):1–8.
- Zhang L, Daizong LIN, Xinyuanyuan SUN, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368(6489):409–412.
- Yoshino R, Yasuo N, Et Sekijima M. Identification of key interactions between SARS-CoV-2 main protease and inhibitor drug candidates. Sci Rep. 2020;10(1):1–8.
- Sourav DAS, Sarmah S, Lyndem S, et al. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J Biomol Struct Dyn. 2021;39(9):3347–3357.
- Hong-zhi DU, Xiao-ying HOU, Yu-huan MIAO, et al. Traditional Chinese medicine: an effective treatment for 2019 novel coronavirus pneumonia (NCP). Chin J Nat Med. 2020;18(3):206–210.
- Benarba BEPANDIELLA, Atanasio. Medicinal plants as sources of active molecules against COVID-19. Front Pharmacol. 2020;11:1189.
- Seri JO, Suwon KIM, Shin DH, et al. Inhibition of SARS-CoV 3CL protease by flavonoids. J Enzyme Inhib Med Chem. 2020;35(1):145–151.