
ABSTRACT
Listeria monocytogenes is a common foodborne pathogen that presents in various food products, posing important threat to public health. The aim of this study was to establish a rapid and sensitive method with visualization to detect L. monocytogenes based on polymerase spiral reaction (PSR). Primers targeting conserved hlyA gene sequence of L. monocytogenes were designed based on bioinformatics analyses on the current available L. monocytogenes genomes. The isothermal amplification PSR can be completed under constant temperature (65ᵒC) within 60 min with high specificity and sensitivity. Twenty-five reference strains were used to evaluate the specificity of the developed reaction. The results showed that the sensitive of the reaction for L. monocytogenes in purified genomic DNA and artificially contaminated food samples were 41 pg/μL and 103 CFU/mL, respectively. It was 100-fold more sensitive than conventional PCR. In conclusion, this novel PSR method is rapid, cost-efficient, timesaving, and applicable on artificially contaminated food samples, providing broad prospects into the detection of foodborne microbes with the promising on-site inspection.
Graphical abstract
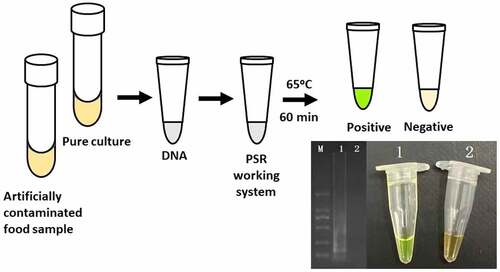
Introduction
Food-borne disease caused by harmful microorganisms is the one of most important food safety issues effecting people’s life. Thousands of incidents of food poisoning occur each year caused by the food-borne pathogens [Citation1]. According to the World Health Organization (WHO), more than 150 million food safety cases happen each year with 175,000 deaths in Southeast Asia which is higher than any other regions. Seventy-seven million people get illness caused by the contaminated food in America which come next to last [Citation2]. Since the first isolation and description of L. monocytogenes in 1926, it has been shown the strong prevalence all over the world. Listeriosis is a food-borne disease among human and animals caused by L. monocytogenes with serious symptoms [Citation3]. Foodborne listeriosis is important contributed by its severe clinical symptoms and high mortality rate of 30% among susceptible group, such as immunocompromised people, pregnant women, babies and the elders [Citation4,Citation5]. L. monocytogenes is able to present both in raw material and food processing.
With the increased attention to the food safety, the development of a rapid detection with visualization for identification of L. monocytogenes is significant and necessary.
Traditionally, the routine detection method of L. monocytogenes requires complicated operation and longtime involving the process of enrichment, colony formation and series of confirmation tests [Citation6]. Except the conventional culture medium, there are detection methods based on the amplification of nucleic acid, including PCR and real-time PCR assays which developed well for the past decades [Citation7]. However, L. monocytogenes detection and/or quantification in foods is still based on culture-dependent methods. Recently, researchers have raised much attention to novel isothermal amplification techniques due to the simple protocols rapid analysis and better performance compared with PCR [Citation8,Citation9]. Polymerase spiral reaction (PSR) is the one of novel isothermal amplification assays developed by Liu et al with efficient amplification of nucleic acid. The main primers Ft and Bt of PSR are composed of two segments, with a sequence between upstream and downstream is added to the front end of upstream primer F and downstream primer B of PCR, respectively. In the presence of Bst DNA polymerase and betaine, the upstream primers Ft and Bt bind to and extend the target sequence, respectively. After dissociation, each single chain contains the sequence we added and its complementary sequence, so it can be folded into a ring structure and continue to extend. The resulting new extension products can also be used as templates for reaction. Repeated primers combine, extend, unchain, single chain rotation and extended cycle will eventually form a series of complex structures with different molecular weight to achieve the purpose of nucleic acid amplification under isothermal conditions. This amplification can be completed under constant temperature with the advantages of simplicity, rapidity, high sensitivity and cost-efficient [Citation10]. The expensive heating machine can be replaced by the simple water bath or other heating block. The whole amplification procedure can be completed within one hour by one pair of primer which span three distinct sequences of a target gene. The products of PSR can be measured based on the turbidity, electrophoresis of amplicons and DNA-specific fluorescent reaction in tubes, such as SYBR Green-I [Citation11]. However, the application of PSR method on detection of L.monocytogenes especially in food samples has not been reported.
Therefore, the current study aimed to establish a rapid and cost-efficient assay to detect the L. monocytogenes by PSR technique based on hlyA gene which might have a contribution on solving food-borne outbreaks related to L. monocytogenes. Additionally, the limit of detection (LOD) and specificity were compared with the PCR assays. The applicability of the PSR in the food industry were evaluated by analysis of food samples.
2 Materials and methods
2.1 Bacteria strains
To standardize and evaluate the reaction system of PSR assay, a total of 25 bacteria strains were used in this study, including five standard strains and 20 non-target strains (). L.monocytogenes ATCC19116, ATCC19114, ATCC19115, ATCC15313, ATCC19113 were used as positive control. All strains used in this study had been preliminarily identified in the Lab of Clinical Microbiology, Zhongshan Supervision Testing Institute of Quality & Metrology. The strains were grown overnight in trypticase soy broth (TSB) at 37ᵒC with shaking at 200 rpm upon further use.
Table 1. Bacterial strains used in this study
2.2 DNA extraction
Pure strains of all bacteria were used for preparation of genomic DNA after incubation in TSB(Huankai Microbial, China) overnight. All genomic DNA were isolated by DNA extraction Kit (Dongsheng Biotech, Guangzhou) according to the manufacturer’s instructions with quantity and quality measured using a Nano Drop 2000 (Thermo Fisher Scientific Inc, Waltham, MA, USA). The isolated DNA was stored at −20ᵒC for further usage.
2.3 Primer design
All current available L. monocytogenes genomes in GenBank database from NCBI were collected and comparative genomic analyses were performed for primer selection. hlyA gene is used in this study as detecting target. Only a single copy of this gene is present in the genome of pathogenic L. monocytogenes. This gene encoding listeriolysin O toxin which is necessary for virulence and is thus used for identifying L. monocytogenes in the presence of other Listeria strains. The primers were designed to identify the specific target hlyA of L. monocytogenes to detect the target and non-target microbes. The protocol of primer design was described as follows. The spiral primers (Ft and Bt) targeting the hlyA gene includea forward and reverse primer [Citation10]. Primers used in this study were listed in , which were designed using Primer Premier 5.
Table 2. Primers sequence used for PSR
2.4 Establishment of PSR assays
Twenty-five reference stains were used to develop and evaluate the specificity and sensitivity of PSR methods. The process of pure culture and DNA extraction were performed as described previously. The final volume of reaction system was 26 μL. The working system includes 20.0 mM Tris-HCl, 10.0 mM, (NH4)2SO4, 10 mM KCl, 8.0 mM, MgSO4, 0.1%, Tween 20, 0.7 M betaine (Sigma, USA), 1.4 mM of dNTP mix, 8 U of Bst DNA polymerase large fragment (NEB, USA), 1.6 μM (each) of the primers Ft and Bt, the specified amounts of genomic DNA and 1 μL mixture chromogenic agent. The total volume of reaction system was made up to 26 μL with nuclease free water. The reaction mixtures of PSR were heated at 65ᵒC for 60 min and terminated at 80ᵒC for 2 min. Nuclease free water instead of DNA template was used as blank control. The amplified products were analyzed by electrophoresis on 1.5% agarose gels.
Ratio of calcein and Mn2+, dosage of betaine and reaction time were compared to optimize the PSR condition. To investigate the effect of dye on the reaction, the ratio of calcein and Mn2+ from 1:20 to 1:2 were tested respectively. Simultaneously, betaine plays a significant role in DNA melting, it is necessary to confirm its concentration in PSR reaction. Moreover, to investigate the minimum reaction time required in a PSR run, 12 different time points had been studied, including 5 min, 10 min, 15 min, 20 min, 25 min, 30 min, 35 min, 40 min, 45 min, 50 min, 55 min and 60 min, respectively.
2.5 Specificity and sensitivity of PSR assay
The specificity of PSR assay was evaluated by the amplification of the genomic DNA extracted from five standard L. monocytogenes and 20 non-L. monocytogenes strains as listed in . Non-target bacteria include Escherichia coli, Salmonella, Staphylococcus aureus, Vibrio parahaemolyticus, Lactobacillus casei and Pseudomonas aeruginosa. Nuclease free water was added as blank control. The reaction of PSR was conducted under the corresponding conditions mentioned above.
To evaluate the sensitivity of the PSR assay, the genomic DNA of L. monocytogenes were serially 10-fold diluted by nuclease free water. The products of PSR were detected by 1.5% agarose gel electrophoresis. All the tests were performed in triplicate.
2.6 Application of PSR assay in rice products
The Application of PSR assay of detection for L. monocytogenes were conducted in rice products (Cantonese rice cake, steamed bread and rice noodle from Guangzhou Restaurant, Guangzhou). Twenty-five gram of frozen pastry (Guangzhou Restaurant, Guangzhou) was added to 225 mL of 0.9% NaCl which were sterilized as food samples and contaminated by L. monocytogenes strains. The concentrations of L. monocytogenes ATCC19113 DNA, extracted from serially diluted contaminated food samples, ranging from 108–10 CFU/mL were subjected to PSR and PCR methods in triplicate [Citation11].
3 Results
3.1 Establishment of PSR method for L. monocytogenes
The PSR assays for hlyA were established using L. monocytogenes ATCC19113. The amplified products were detected by 1.5% agarose gel electrophoresis, and the bands were observed under UV light (). Agarose gel analysis revealing the typical electrophoresis pattern of PSR amplified product, which is not a single band but a ladder pattern because the PSR method forms amplified products of various sizes consisting of alternately inverted repeats of the target sequence on the same strand. In addition, 1 μL mixture chromogenic agent (MgCl2 and calcein) we added into the PSR reaction system, wherein the dye color simultaneously changed from original to green in the positive sample, or water retained the original orange color ().
Figure 1. Results of the PSR assays for detection invA gene; (a) observation of amplification products with 1.5% agarose gel electrophoresis under UV light and fluorescence dye by naked eye (b); M, DNA marker; lane 1, negative control; lane 2, positive products (a); tube 1, positive products; tube 2, negative control.

3.2 Specificity and sensitivity of PSR assay and its application
The PSR assay was evaluated for its specificity using five L. monocytogenes strains and 20 non-L. monocytogenes reference strains as control. Of the 25 strains, only the DNA of L. monocytogenes strains were amplified successfully. No cross-reaction was found with all the related non-target strains indicating the high specificity of the designed PSR primers (). The analytical sensitivity of the PSR assay was measured using 10-fold serial dilutions of genomic DNA of L. monocytogenes ATCC19113. After amplification reaction, the results revealed that the LOD of PSR reaction was 41 pg/μL for hlyA (). While that of the PCR was 4.1 ng/μL, indicating that the sensitivity of PSR assay was significantly higher than the traditional PCR method.
Figure 2. Specificity of PSR assay for detection L. monocytogenes strains with hylA genes by 1.5% agarose gel electrophoresis; M-DNA marker; lane 1–5, L. monocytogenes ATCC19116, ATCC19114, ATCC19115, ATCC15313, ATCC19113; lane/tube 6–25, non-L. monocytogenes strains of Escherichia coli O157:H7 (ATCC43894, ATCC43895, E019, E020, E043, E044), Salmonella enteric (ATCC29629, ATCC14028, ATCC19585, ATCC13076), Vibrio parahaemolyticus (ATCC27969, ATCC17802), Pseudomonas aeruginosa (ATCC27853, C9, C40), Staphylococcus aureus (ATCC23235, ATCC25923, 10085, 10071), and Lactobacillus casei; lane 26, negative control.
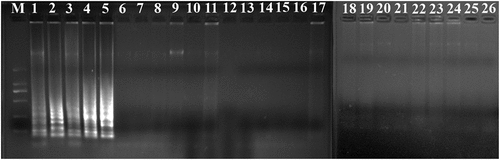
Figure 3. Sensitivity of the PSR assay in genomic DNA of L. monocytogenes with hylA genes by 1.5% agarose gel electrophoresis (a) and fluorescence dye by naked eye (b); M-DNA marker; lane/tube 1–8, 41 ng/μL, 4.1 ng/μL, 410 pg/μL, 41 pg/μL, 4.1 pg/μL, 410 fg/μL, 4.1 fg/μL, Negative control.

3.3 Application of detect L. monocytogenes in rice products
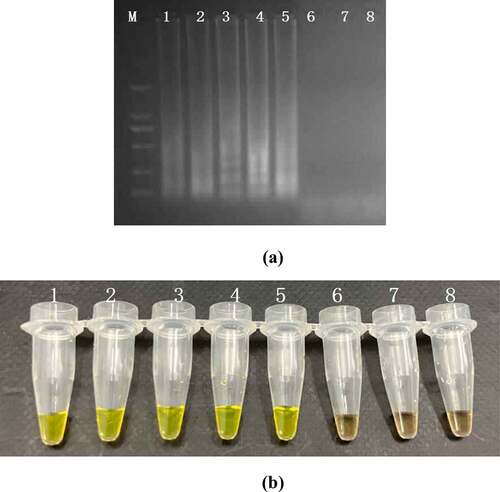
The sensitivity of PSR and PCR assay were performed by a series 10-fold diluted concentration (108–10 CFU/mL) of DNA templates which were extracted from the artificially contaminated rice products. The results were subject to detect by electrophoresis in 1.5% agarose gels and fluorescence dye by naked eye (). The sensitivity of PSR assay for hlyA gene in food samples were 103 CFU/mL and no false positive amplification was observed, indicating high specificity of the established PSR assays.
Figure 4. Sensitivity of the PSR assay in genomic DNA of L. monocytogenes with hylA genes from food samples: by 1.5% agarose gel electrophoresis (a) and fluorescence dye by naked eye (b); M-DNA marker; lane/tube 1–8, 107 CFU/mL; 106 CFU/mL; 105 CFU/ mL; 104 CFU/mL; 103 CFU/mL; 102 CFU/mL; 101 CFU/mL, Negative control.
4 Discussion
L. monocytogenes is an important food-borne pathogen around the world due to their resistance to antibiotics and adverse conditions, such as low pH, temperature and high salt concentration. It is found in various food commonly, especially fish, meat, poultry and dairy products [Citation12]. Thus, the development of a rapid and intuitive detection method for this food-borne microbe is essential from the perspective of public health.
Normally, the confirmation of amplification reaction would be performed by the electrophoresis which is easier to cause the false positive results due to the process of open reaction cover. However, with the improvement of fluorescence reaction, results can be identified by the color change of reaction tube by naked eye which saves much time for whole detection. In current study, the entire reaction can be achieved within 60 min without the requirement of sophisticated equipment. This innovative isothermal amplification, PSR, has been used to detect various microbes, including E. coli, Salmonella, P. aeruginosa, Candida albicans and V. parahaemolyticus [Citation10,Citation11,Citation13–15]. The PSR technique developed in this study can be specific to detect L. monocytogenes strains both in pure culture and food samples with 100 times higher sensitivity than PCR.
5 Conclusion
In conclusion, we have demonstrated that PSR assays could be useful and powerful tools for the rapid detection of L. monocytogenes by targeting on hlyA gene. Undoubtedly, the simplicity, rapidity and sensitivity of PSR assay make it an ideal routine diagnostic tool for rapid diagnosis of food-borne pathogens in both commercial and clinical fields that will strongly support the cooperation of patients and clinicians in endemic areas. With such advantages, the established PSR assay is a potentially highly complementary method to cultural approaches for quick and accurate detection of L. monocytogenes in foodstuff tested as part of outbreak investigations and other surveillance projects.
Highlights
The isothermal amplification method PSR can be completed under constant temperature within 60 min with high specificity and sensitivity.
The developed PSR assay is 100-fold more sensitive than conventional PCR in the detection of L. monocytogenes.
The PSR assay is applicable in the detection of L. monocytogenes in artificially contaminated food samples.
Acknowledgements
This work was supported by the State Key Laboratory of Applied Microbiology Southern China (Grant No. SKLAM005-2019), the Open Project of the State Key Laboratory of Respiratory Disease (GHMJLRID-Z-202118), National Natural Science Foundation of China [82170056], the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program [2017BT01S155], National key Research and Development program of China [2018YFC1311600, 2018YFC1311604, 2016YFC1304100, 2016YFC1304104], Natural Science Foundation of Guangdong (2021A1515011024), Science and Technology Program of Guangzhou (202102080045), The Doctoral Fund of the Third Affiliated Hospital of Guangzhou Medical University (2019B07), Open Project of the State Key Laboratory of Respiratory Disease (SKLRD-Z-202103), Characteristic Innovation Projects of Universities in Guangdong Province (2019KTSCX139), Guangdong Major Project of Basic and Applied Basic Research (2020B0301030005), Guangdong International S&T Cooperation Programme (2021A0505030007).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Wang L, Li Y, Chu J, et al. Development and application of a simple loop-mediated isothermal amplification method on rapid detection of Listeria monocytogenes strains. Mol Biol Rep. 2012;39(1):445–449.
- Davies OL, Chaib F. WHO’s first ever global estimates of foodborne diseases find children under 5 account for almost one third of deaths. 2015. [cited 2015 Dec 3]. Available from: https://www.who.int/news-room/detail/03-12-2015-who-s-first-ever-global-estimates-of-foodborne-diseases-find-children-under-5-account-for-almost-one-third-of-deaths
- Low JC, Donachie W. A review of Listeria monocytogenes and listeriosis. Vet J. 1997;153(1):9–29.
- Sutherland PS, Porrit RJ. Food-borne microorganisms of public health significance. Hocking AD, ed. North Sydney: AIFST (NSW Branch), Food Microbiology Group; 1997. p. 333–378.
- Swaminathan B, Gerner-Smidt P. The epidemiology of human listeriosis. Microbes Infect. 2007;9(10):1236–1243.
- Amagliani G, Omiccioli E, Campo A, et al. Development of a magnetic capture hybridization-PCR assay for Listeria monocytogenes direct detection in milk samples. J Appl Microbiol. 2006;100(2):375–383.
- Simon MC, Gray DI, Cook N. DNA Extraction and PCR methods for the detection of listeria monocytogenes in cold-smoked salmon. Appl Environ Microbiol. 1996;62(3):822–824.
- Dean FB, Nelson JR, Giesler TL, et al. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001;11(6):1095–1099.
- Euler M, Wang Y, Otto P, et al. Recombinase polymerase amplification assay for rapid detection of Francisella tularensis. J Clin Microbiol. 2012;50(7):2234–2238.
- Liu W, Dong D, Yang Z, et al. Polymerase Spiral Reaction (PSR): a novel isothermal nucleic acid amplification method. Sci Rep. 2015;5(1):12723.
- Xu W, Gao J, Zheng H, et al. Establishment and application of polymerase spiral reaction amplification for salmonella detection in food. J Microbiol Biotechnol. 2019;29(10):1543–1552.
- Gao W, Huang H, Zhang Y, et al. Recombinase polymerase amplification-based assay for rapid detection of listeria monocytogenes in food samples. Food Analytical Methods. 2016;10(6):1972–1981.
- Dong D, Zou D, Liu H, et al. Rapid detection of Pseudomonas aeruginosa targeting the toxA gene in intensive care unit patients from Beijing, China. Front Microbiol. 2015;6:1100.
- He S, Jang H, Zhao C, et al. Rapid visualized isothermal nucleic acid testing of Vibrio parahaemolyticus by polymerase spiral reaction. Anal Bioanal Chem. 2020;412(1):93–101.
- Jiang X, Dong D, Bian L, et al. Rapid detection of Candida albicans by polymerase spiral reaction assay in clinical blood samples. Front Microbiol. 2016;7:916.