
ABSTRACT
Previous studies showed dexmedetomidine (DEX) could alleviate myocardial ischemia/reperfusion injury (MIRI). Nevertheless, the mechanisms by which DEX alleviated MIRI remain to be determined. Our results demonstrated that DEX reversed hypoxia/reoxygenation (H/R)-induced decreased proliferation, and enhanced LINC00982 level, apoptosis, and inflammation in H9c2 cells. Moreover, LINC00982 overexpression attenuated the DEX-mediated protective effect of H9c2 cells under H/R. In addition, DEX upregulated p-phosphoinositide-3-kinase (p-PI3K) and p-protein kinase B (p-AKT) levels, and the silencing of LINC00982 further enhanced this effect in H/R-induced H9c2 cells. Furthermore, LINC00982 deletion enhanced the protective effect of DEX on H9c2 cells under H/R condition, while PI3K inhibitor, LY294002, obviously reversed this phenomenon. In sum, our work determined that DEX could suppress cell apoptosis and inflammation in H/R-triggered H9c2 through downregulating LINC00982 and activating PI3K/AKT signaling.
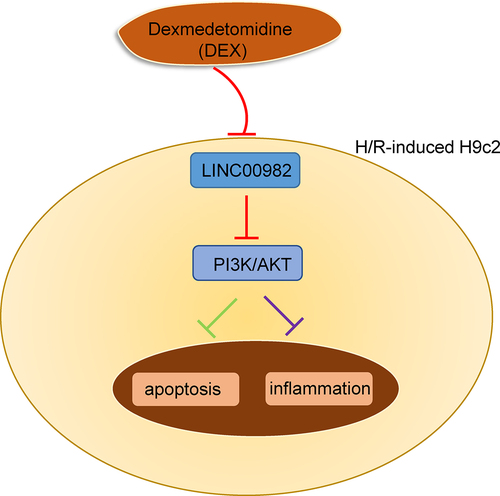
Graphical abstract
Introduction
Acute myocardial infarction (AMI), characterized by a reduced or complete cessation of blood flow to the heart, is a major threat to human health and results in high mortality worldwide [Citation1]. At present, rapid reperfusion is the main treatment strategy for AMI, but they may result in cardiomyocyte dysfunction, such as myocardial ischemia/reperfusion injury (MIRI) [Citation2], and the mortality rate of MIRI is approximately 10% among AMI patients who received rapid reperfusion therapy [Citation3]. Therefore, it is necessary to elucidate mechanisms underlying MIRI and search for novel targets to inhibit reperfusion injury and protect cardiac function.
Dexmedetomidine (DEX) is a highly selective α 2-adrenergic receptor agonist that is extensively used in clinical anesthesia and intensive care unit, exerting sedative, analgesic, and sympatholytic effects [Citation4]. Previous studies have demonstrated that DEX exerted its cardioprotective effect on MIRI via diverse regulatory pathways. For example, DEX reduced lncRNA MALAT1 expression to improve MIRI through elevating miR-346 level [Citation5]. DEX alleviated MIRI in rats through PI3K/AKT/GSK-3β pathway [Citation6]. However, the specific mechanism by which DEX alleviated MIRI remains to be elucidated.
Long noncoding RNA (lncRNA) is a type of RNA that is >200 nt without an open reading frame or coding sequences, which takes part in various biological processes, including differentiation, proliferation, angiogenesis, and metastasis [Citation7–9]. Increasing evidence indicated that lncRNA acted as a vital regulator in the development of MIRI. For instance, lncRNA MALAT1 blocking could inhibit MIRI and improve the cardiac function of rats via activation of the AKT signaling [Citation10]. Overexpression of lncRNA FGD5-AS1 relieved MIRI through inhibition of miR-106a-5p and miR-106b-5p expression [Citation11]. Besides, Abhd11os deletion attenuated MIRI through repressing apoptosis of cardiomyocytes [Citation12]. A research from Zhang et al implied that LINC00982 overexpression restrained cell viability and facilitated apoptosis via modulating the PI3K/AKT pathway [Citation13]. Nonetheless, the role and function of LINC00982 in MIRI are still unknown.
Herein, we hypothesized that DEX could exert its protective effects against MIRI, and we aimed to explore the underlying molecular mechanism of DEX in MIRI. Our study demonstrated that DEX promoted cell proliferation and inhibited cell apoptosis by regulating LINC00982 and activating the PI3K/AKT signaling in H/R-induced H9c2 cells. These findings might provide a theoretical basis for the clinical application of DEX in the treatment of MIRI.
Materials and methods
Hypoxia/reoxygenation (H/R) model
The cardiomyocyte H9c2 cells were purchased from BeNa culture collection (Beijing, China) and cultured in DMEM containing 10% FBS, 100 U/ml penicillin and 0.1 mg/ml streptomycin in an incubator (95% air+5% CO2) at 37°C. To establish a H/R cell model to mimic MIRI, the cells were placed in the hypoxia condition (1% O2, 5% CO2, and 94% N2) for 24 h, followed by reoxygenation in a normoxic chamber (5% CO2 and 95% air) for 6 h. The H9c2 cells in Dex groups were subjected to 10 nM Dex pretreatment for 1 h before H/R insult.
Cell transfection
The specific short hairpin RNAs (shRNAs) targeting LINC00982 (shLINC00982; 5’- GCAATAAACGGAATCGGTTTC-3’) with its negative control (shNC), and pcDNA3.1/LINC00982 with its negative control (pcDNA3.1), obtained from GenePharma (Shanghai), were transfected into H9c2 cells using Lipofectamine 2000 (Invitrogen, USA).
RT-qPCR
Total RNAs were isolated from different treated cells by Trizol (Invitrogen). rime-Script TMRT reagent kit (Takara) was used to reversely transcribed RNA into cDNA. qPCR was conducted using SYBR Green PCR master mix on a 7500 Real-Time PCR System. Primers used for targets amplification were presented in .
Table 1. Primer sequences for RT-qPCR
MTT assay
H9c2 cells were seeded into 96-well plate and treated with 10 µl MTT solution for 24 h. Then, 100 µl DMSO (Sigma-Aldrich) was added to each well and the plates were gently agitated for 10 min. The absorbance was determined at 490 nm using a microplate reader (BioTek) [Citation14].
Enzyme-linked immunosorbent assay (ELISA)
IL-1β, IL-6, and TNF-α in the supernatants of H9c2 cells were measured by the corresponding ELISA kit (Invitrogen, USA), following the manufacturer’s instructions [Citation15,Citation16].
Flow cytometry
An Annexin V-FITC/PI Apoptosis Detection Kit (BD Biosciences) was applied to analyze cardiomyocyte apoptosis. Briefly, H9c2 cells were re-suspended in 100 μL binding buffer, stained with Annexin V-FITC and PI reagent, and then cultured in darkness for 10 min. The apoptotic rate was evaluated by flow cytometry.
Western blotting
The cultured or transfected cells were lysed in RIPA buffer ((Beyotime) with 1% PMSF. The proteins were separated by 10% SDS-PAGE and transferred to PVDF membranes. Then, the membranes were incubated with the following primary antibodies: Bax (ab32504, Abcam), cleaved-caspase-3 (ab2302, Abcam), p-AKT (Ser473, #4060, Cell Signaling Technology), AKT (#4691, Cell Signaling Technology), p-PI3K (Tyr458, #17,366, Cell Signaling Technology), PI3K (#4249, Cell Signaling Technology), and GAPDH (ab8245, Abcam) at 4°C overnight. Subsequently, the corresponding secondary antibody was added for incubation. Finally, ECL System was employed to detect the protein expression.
Statistical analysis
GraphPad 6.0 software was used for the statistical analyses and data are presented as the mean ± SD. Comparisons between groups were analyzed using one-way ANOVA followed by Tukey’s post hoc test. p <0.05 indicated statistical significance.
Results
In this study, we attempted to investigate the function and molecular mechanism of DEX in H/R-challenged cardiomyocyte dysfunction. The results manifested that DEX protected H9c2 cells against H/R injury via regulating LINC00982 and activating PI3K/AKT pathway, providing a scientific basis for DEX application in MIRI.
DEX decreases LINC00982 expression and suppresses the apoptosis and inflammation in H9c2 cells under H/R conditions
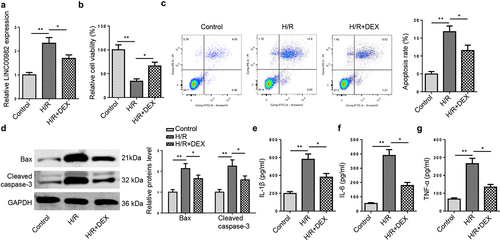
To determine the role of DEX in MIRI, an in vitro MIRI cell model was constructed. RT-qPCR indicated that LINC00982 was notably upregulated in H/R-stimulated H9c2 cells, while such phenomenon was restored following DEX treatment ()). In addition, H/R treatment impeded cell viability, which was reversed by DEX ()). Furthermore, flow cytometry implied that H/R dramatically accelerated H9c2 cell apoptosis, while the promotive impact was abrogated by DEX treatment ()). Consistently, H/R obviously lifted apoptosis-related biomarkers, including Bax and cleaved caspase-3 levels, while this effect was restored by DEX treatment in H9c2 cells ()). Besides, H/R elevated levels of IL-1β, IL-6 and TNF-α in H9c2 cells, which was inhibited by DEX ()).
Figure 1. DEX decreases LINC00982 expression and suppresses the apoptosis and inflammation in H9c2 cells under H/R conditions. (a) RT-qPCR showed LINC00982 expression in H9c2 cells after H/R and DEX treatment. (b) Cell viability was measured by MTT. (c) Cell apoptosis rate was measured by flow cytometry. (d) Western blot showed levels of Bax and cleaved caspase-3. (e-g) The levels of proinflammatory cytokines were measured by ELISA.
DEX exerts its protective effects against H/R-induced H9C2 cell damage by regulating LINC00982
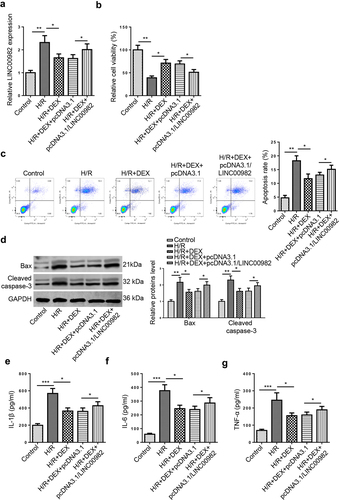
To investigate whether DEX exerted protective effects on MIRI by regulating LINC00982, H9c2 cells were treated with DEX without or with LINC00982 overexpression plasmid under H/R conditions. RT-qPCR analysis determined that DEX treatment inhibited H/R-induced upregulation of LINC00982, while LINC00982 augmentation rescued this change ()). Then, the result from MTT displayed that DEX promoted the cell viability of H9c2 cells, while LINC00982 overexpression reversed this effect ()). On the contrary, LINC00982 addition abrogated the suppressive impact of DEX on apoptosis of H9c2 cells under H/R ()). Moreover, western blot revealed that LINC00982 overexpression neutralized the inhibition of Bax and cleaved caspase-3 levels induced by DEX ()). Meanwhile, ELISA determined that LINC00982 overexpression restored the anti-inflammatory effect of Dex on H9c2 cells ()). All these data demonstrated that the impacts of DEX on MIRI were reversed by LINC00982 supplementation.
Figure 2. DEX exerts its protective effects on H/R-induced H9C2 cells by regulating LINC00982. H9c2 cells were treated with DEX, DEX+pcDNA3.1, or DEX+pcDNA3.1/LINC00982 under H/R conditions. (a) RT-qPCR showed LINC00982 expression in different groups. (b) Cell viability in different groups was measured using MTT assay. (c) Cell apoptosis rate was measured using flow cytometry. (d) Western blotting showed protein levels of apoptosis-related factors (Bax and cleaved caspase-3) in different groups. (e-g) The levels of proinflammatory cytokines were determined by ELISA.
DEX reverses H/R-induced inactivation of PI3K/AKT pathway by downregulating LINC00982
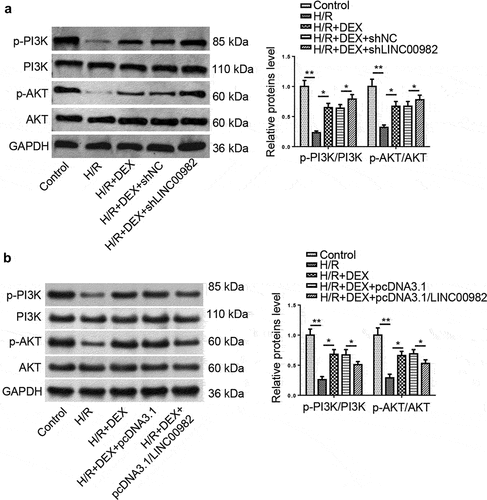
In previous studies, PI3K/AKT signaling has been shown to act as a crucial function in I/R injury [Citation17]. Herein, we wonder whether PI3K/AKT signaling was implicated in the DEX-mediated protection against MIRI. As displayed in ), DEX reversed the downregulated phosphorylation levels of PI3K and AKT induced by H/R, and this pathway was activated or inactivated by LINC00982 knockdown or overexpression. In sum, DEX rescued the inactivation of PI3K/AKT pathway caused by H/R via regulating LINC00982.
Figure 3. DEX reverses H/R-induced inactivation of PI3K/AKT pathway by downregulating LINC00982. (a and b) The levels of p-PI3K and p-AKT in H9c2 cells treated with DEX, DEX+shNC, DEX+shLINC00982, DEX+pcDNA3.1, or DEX+pcDNA3.1/ LINC00982 under H/R conditions were evaluated in by western blotting.
DEX improves H/R-induced MIRI by regulating LINC00982 and activating PI3K/AKT pathway
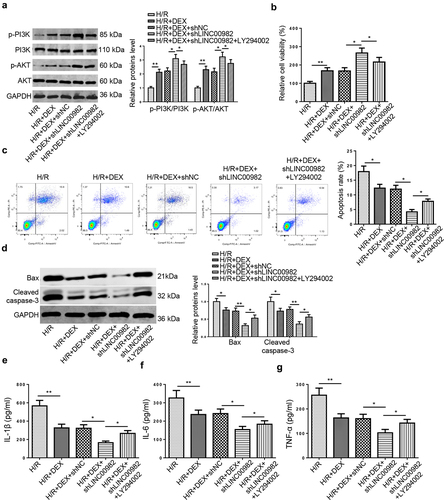
To further confirm whether DEX mediated H/R-induced MIRI via regulating PI3K/AKT pathway, LY294002 (a PI3K inhibitor) was used to inactive PI3K/AKT signaling. Firstly, the results from RT-qPCR manifested that LY294002 treatment restored the promotive impacts of DEX and LINC00982 silencing on the p-PI3K and p-AKT levels ()). Then, LINC00982 knockdown enhanced DEX-mediated increased cell viability and attenuated apoptosis rate, whereas LY294002 obviously reversed these impacts ()). Moreover, LINC00982 deficiency aggravated the suppressive effects of DEX on apoptosis-related protein and proinflammatory cytokine levels, which was distinctly restored by LY294002 treatment ()). These data indicated that DEX expedited cell viability, reduced the apoptosis and inflammation in H/R-induced cardiomyocytes through regulating LINC00982 and PI3K/AKT pathway.
Figure 4. DEX improves H/R-induced MIRI by regulating LINC00982 and activating PI3K/AKT pathway. H9c2 cells were treated with DEX, DEX+shNC, DEX+shLINC00982, or DEX+shLINC00982+ LY294002 under H/R condition. (a) Western blotting showed protein levels of p-PI3K and p-AKT in different groups. (b) Cell viability in different groups was measured using MTT assay. (c) Cell apoptosis was measured using flow cytometry. (d-g) The levels of apoptosis-related proteins and proinflammatory cytokines were measured.
Discussion
Previous researches indicated that DEX could protect hearts against ischemia/reperfusion damage and myocardial cells from H/R damage [Citation18]. This work offered new insights into the molecular mechanism of DEX in MIRI and provided the first evidence of a link between DEX and LINC00982 and its downstream signaling in the MIRI. Our data indicated that DEX protected H9c2 cells against H/R injury via regulating LINC00982 and activating PI3K/AKT pathway.
Recently, many studies have demonstrated that lncRNAs participated in cardiac ischemia-related cardiovascular diseases [Citation19,Citation20]. For instance, Liu et al reported that SNHG7 deletion repressed H/R-induced cardiomyocyte oxidative stress and apoptosis via increasing miR-181b-5p expression, thereby exerting a protective effect on cardiomyocytes [Citation21]. Mo et al displayed that lncRNA CHRF was upregulated in MIRI, and CHRF silencing could relieve MIRI by attenuating myocardial cell apoptosis [Citation22]. Xu et al elaborated that LINC00982 addition could promote cell apoptosis through modulating PI3K/AKT signaling in papillary thyroid carcinoma [Citation23]. Niu et al exhibited that deficiency of lncRNA HRIM improved H/R-triggered myocardial damage and repressed inflammatory response by inactivating the NF-κB pathway [Citation24]. Herein, we identified that LINC00982 was upregulated in H9c2 cells under H/R conditions, while DEX declined LINC00982 expression. Moreover, LINC00982 supplementation attenuated the protective impact of DEX on H/R-triggered H9c2 cells. Thus, DEX could improve H/R-induced H9c2 injury by downregulating LINC00982.
The PI3K/AKT pathway is associated with numerous cellular processes, such as inflammation response, cell viability, apoptosis, and survival [Citation25]. Moreover, accumulating studies have identified the protective effect of the PI3K/AKT pathway on MIRI [Citation26]. For instance, Yao et al revealed that insulin-induced PI3K/AKT signaling activation inhibited cardiomyocyte apoptosis and could improve cardiac function [Citation26]. Shen et al manifested that Sulodexide inhibited endoplasmic reticulum stress caused by MI/R by activating the PI3K/AKT signaling [Citation27]. Zhang et al elucidated that Nobiletin improved MIRI through suppressing endoplasmic reticulum stress-related apoptosis by modulating the PI3K/AKT signal pathway [Citation28]. These previous studies provided evidence that activated PI3K/AKT signaling may improve MIRI. In our work, DEX upregulated p-PI3K and p-AKT expression, and the downregulation of LINC00982 enhanced the activation of this signaling in H9c2 cells under H/R conditions. In addition, LINC00982 silence enhanced the protective effects of DEX on H/R-treated H9c2 cells, whereas PI3K inhibitor restored these effects.
Conclusions
We demonstrated that DEX improved MIRI by regulating LINC00982 and activating PI3K/AKT signaling. Our study elucidated the molecular mechanism underlying the cardioprotective effect of DEX on MIRI and might provide a scientific basis for DEX application in MIRI.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Sun SJ, Wu XP, Song HL, et al. Baicalin ameliorates isoproterenol-induced acute myocardial infarction through iNOS, inflammation, oxidative stress and P38MAPK pathway in rat. Int J Clin Exp Med. 2015;8:22063–22072.
- Gerczuk PZ, Kloner RA. An update on cardioprotection: a review of the latest adjunctive therapies to limit myocardial infarction size in clinical trials. J Am Coll Cardiol. 2012;59:969–978.
- Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet. 2003;361:13–20.
- Hsu YW, Cortinez LI, Robertson KM, et al. Dexmedetomidine pharmacodynamics: part I: crossover comparison of the respiratory effects of dexmedetomidine and remifentanil in healthy volunteers. Anesthesiology. 2004;101:1066–1076.
- Xie MY, Hou LJ. Dexmedetomidine down-regulates lncRNA MALAT1 to attenuate myocardial ischemia reperfusion-induced injury by increasing miR-346. Int J Cardiol. 2021;334:104.
- Zhang X, Xu M, Che X, et al. Dexmedetomidine reduces myocardial ischemia-reperfusion injury in rats through PI3K/AKT/GSK-3beta signaling pathway. Minerva Cardioangiol. 2020;68:58–59.
- Kumar MM, Goyal R. LncRNA as a therapeutic target for angiogenesis. Curr Top Med Chem. 2017;17:1750–1757.
- Hou J, Zhou C, Long H, et al. Long noncoding RNAs: novel molecules in cardiovascular biology, disease and regeneration. Exp Mol Pathol. 2016;100:493–501.
- Shen Y, Gao X, Tan W, et al. STAT1-mediated upregulation of lncRNA LINC00174 functions a ceRNA for miR-1910-3p to facilitate colorectal carcinoma progression through regulation of TAZ. Gene. 2018;666:64–71.
- Sun T, Cheng YT, Yan LX, et al. LncRNA MALAT1 knockdown alleviates myocardial apoptosis in rats with myocardial ischemia-reperfusion through activating PI3K/AKT signaling pathway. Eur Rev Med Pharmacol Sci. 2019;23:10523–10531.
- Hao L, Wang J, Bi SJ, et al. Upregulation of long noncoding RNA FGD5-AS1 Ameliorates myocardial ischemia/Reperfusion injury via MicroRNA-106a-5p and MicroRNA-106b-5p. J Cardiovasc Pharmacol. 2021;78:e45–e54.
- Liu X, Hua Y, Hu M, et al. Knockdown of lncRNA Abhd11os attenuates myocardial ischemia/reperfusion injury by inhibiting apoptosis in cardiomyocytes. J Cardiovasc Pharmacol. 2021. DOI:10.1097/FJC.0000000000001074.
- Zhang C, Li XY, Luo ZZ, et al. Upregulation of LINC00982 inhibits cell proliferation and promotes cell apoptosis by regulating the activity of PI3K/AKT signaling pathway in renal cancer. Eur Rev Med Pharmacol Sci. 2019;23:1443–1450.
- Hao W, Lin F, Shi H, et al. Long non-coding RNA OIP5-AS1 regulates smoke-related chronic obstructive pulmonary disease via targeting micro RNA −410-3p/IL-13. Bioengineered. 2021;12:11664–11676.
- Liu J, Jiang M, Deng S, et al. miR-93-5p-containing exosomes treatment attenuates acute myocardial infarction-induced myocardial damage. Molecular Therapy - Nucleic Acids. 2018;11:103–115.
- Zhang Y, Zhang Y, Wang S, et al. SP1-induced lncRNA ZFPM2 antisense RNA 1 (ZFPM2-AS1) aggravates glioma progression via the miR-515-5p/Superoxide dismutase 2 (SOD2) axis. Bioengineered. 2021;12:2299–2310.
- Vilahur G, Gutierrez M, Casani L, et al. P2Y12 antagonists and cardiac repair post-myocardial infarction: global and regional heart function analysis and molecular assessments in pigs. Cardiovasc Res. 2018;114:1860–1870.
- Song J, Du J, Tan X, et al. Dexmedetomidine protects the heart against ischemia reperfusion injury via regulation of the bradykinin receptors. Eur J Pharmacol. 2021;911:174493.
- Huang Y. Exosomal lncRNAs from mesenchymal stem cells as the novel modulators to cardiovascular disease. Stem Cell Res Ther. 2020;11:315.
- Jiang X, Ning Q. The emerging roles of long noncoding RNAs in common cardiovascular diseases. Hypertens Res. 2015;38:375–379.
- Liu Z, Jin W, Han M, et al. [Silencing LncRNA SNHG7 alleviates hypoxia/reoxygenation-induced cardiomyocyte damage by regulating the expression of miR-181b-5p]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2021;38:812–817.
- Mo Y, Wu H, Zheng X, et al. LncRNA CHRF aggravates myocardial ischemia/reperfusion injury by enhancing autophagy via modulation of the miR-182-5p/ATG7 pathway. J Biochem Mol Toxicol. 2021;35:e22709.
- Xu D, Yu J, Zhuang S, et al. Overexpression of long non-coding RNA LINC00982 suppresses cell proliferation and tumor growth of papillary thyroid carcinoma through PI3K-ATK signaling pathway. Biosci Rep. 2019;39. DOI:10.1042/BSR20191210
- Niu L, Zhao Y, Liu S, et al. Silencing of long noncoding RNA HRIM protects against myocardial ischemia/reperfusion injury via inhibition of NFkappaB signaling. Mol Med Rep. 2020;22:5454–5462.
- Huang F, Zhao JL, Wang L, et al. miR-148a-3p mediates notch signaling to promote the differentiation and M1 activation of macrophages. Front Immunol. 2017;8:1327.
- Yao H, Han X, Han X. The cardioprotection of the insulin-mediated PI3K/Akt/mTOR signaling pathway. Am J Cardiovasc Drugs. 2014;14:433–442.
- Shen D, Chen R, Zhang L, et al. Sulodexide attenuates endoplasmic reticulum stress induced by myocardial ischaemia/reperfusion by activating the PI3K/Akt pathway. J Cell Mol Med. 2019;23:5063–5075.
- Zhang BF, Jiang H, Chen J, et al. Nobiletin ameliorates myocardial ischemia and reperfusion injury by attenuating endoplasmic reticulum stress-associated apoptosis through regulation of the PI3K/AKT signal pathway. Int Immunopharmacol. 2019;73:98–107.