
ABSTRACT
Here, we show the strategies to strengthen Mg alloys through modifying the matrix by planar faults and optimizing the local lattice strain by solute atoms. The anomalous shifts of the local phonon density of state of stacking faults (SFs) and long periodic stacking-ordered structures (LPSOs) toward the high-frequency mode are revealed by HCP-FCC transformation, resulting in the increase of vibrational entropy and the decrease of free energy to stabilize the SFs and LPSOs. Through integrating bonding charge density and electronic density of states, electronic redistributions are applied to reveal the electronic basis for the ‘strengthening’ of Mg alloys.
GRAPHICAL ABSTRACT
IMPACT STATEMENT
Through integrating the bonding charge density, the phonon and electronic density of states, this work provides an atomic and electronic insight into the strengthening mechanism of Mg alloys.
1. Introduction
Mg alloys, being the lightest metallic structural materials, are particularly attractive for transportation applications such as automobiles and aircrafts for weight reducing and higher fuel efficiency and biocompatible and biodegradable features ideal for implant applications [Citation1–4]. However, Mg alloys generally suffer from low ductility at low temperatures due to the limited number of active slip systems and the highly anisotropic critical resolved shear stress of different slip systems [Citation5–7]. The improvement of both the strength and the ductility is still a challenge in the development of advanced Mg alloys. Recent works have shown that the fault layers enriched of solute atoms could stabilize the long period stacking-ordered (LPSO) structures described by a common structural unit composed of local FCC-type stacking sequence [Citation8–10] and improve the mechanical properties of Mg alloys [Citation11–14]. For instance, the tensile yield strength and the elongation of an Mg97Zn1Y2 (at. %) alloy with the 6H LPSO structure produced by rapid solidification can reach 610 MPa and 5%, respectively [Citation12]. It is noted that the 6H LPSO is a structure between the growth fault and the deformation fault [Citation10,Citation15], making it attractive to be studied. Due to the contributions of stacking faults (SFs) with nanoscale spacing, the ultimate tensile strength of Mg–8.5Gd–2.3Y–1.8Ag–0.4Zr (wt. %) can reach as high as 600 MPa and an elongation around 5.2% [Citation14]. Since the planar faults (such as the FCC-type bonding layers in SFs and long periodic stacking-ordered structures (LPSOs), ∼1 nm in thickness) affect core structures and mobility of dislocations [Citation16,Citation17], twinnability and ductility [Citation18,Citation19], strengthening [Citation20,Citation21], and creep rate [Citation22], it is essential to understand the atomic and electronic basis for the strengthening and the deformation mechanisms in Mg alloys.
Since the vibrational entropy is proportional to the logarithmic moment of phonon density of states (DOS), lattice vibrations—regulated significantly by point defects (solute atoms or vacancy) and planar defects (SFs or grain boundaries)—play an important role in determining phase stability [Citation23,Citation24]. For example, previous studies have shown the high entropy stabilization of ordered Zn4Sb3 [Citation25] and Ni3Pt [Citation26]. Furthermore, based on the analysis of local phonon DOS (LPDOS) of Σ5(310) grain boundaries in Al, it has been shown that the atomic vibrations of the atoms around the boundaries are different from those far away in the bulk, which could improve the resonant mode at low frequency and weaken the mode at high frequency [Citation27]. Similarly, with the formation of the Σ5(310) grain boundary in Cu, the most striking feature is the increase of the LPDOS at low frequency and its decrease at high frequency. The anomalous shifts of the LPDOS peaks at high-frequency region are also observed in Σ5 grain boundaries of Al and Cu [Citation27,Citation28]. While in the study of lattice stability of SFs and LPSOs of Mg alloys, the contribution of the free energy of harmonic and anharmonic effect on lattice vibration must be discussed, which dominates the thermodynamic properties (i.e. entropy, heat capacity, and free energies) at finite temperatures [Citation29,Citation30].
In this work, we study the contributions of fault layers (included in various SFs and LPSOs) on the phonon DOS and vibrational energies of Mg at finite temperature. The anomalous shifts of the LPDOS of SFs and LPSOs toward high frequency are revealed by the HCP-FCC bond transformation, resulting in the increase of vibrational entropy and the decrease of free energy to stabilize the corresponding structures. Moreover, it is understood that Mg could be strengthened by the formation of FCC-type nano-lamellar fault layers, which are stabilized by the segregation of the solute atoms due to their contributions to the energy, bond structure and strength, and elastic properties. Through integrating the characterizations of bonding charge density (Δρ) and electronic DOS (eDOS), the atomic and the electronic basis for solid solute strengthening mechanism is revealed by electronic redistributions, providing a fundamental insight into the ‘strengthening’ of Mg alloys. This work supports a qualitative description of the strengthening mechanism in the development of advanced Mg alloys.
The rest of the paper is organized as follows. In Section 2, the computational methodologies are presented in detail. In Section 3, the effects of lattice vibrations of SFs and LPSOs of pure Mg on the lattice stabilities are firstly investigated by the LPDOS. Secondly, according to the contributions of solute atoms to stacking fault energy, segregation energy, shear modulus, bulk modulus, ideal shear stress and bond strength, the impact of solute atoms on the partial dislocation width of binary Mg-X alloys is discussed. And then, the bond structure and strength of Mg97.5Gd1.67TM0.83 (TM = Zn and Zr) alloys affected by the solute atoms and the fault layers are discussed. Through integrating the various aspects of pure Mg, binary Mg-X and ternary Mg-Gd-TM, the candidate strategies strengthening Mg alloys are discussed in Section 4. Finally, a summary is presented in Section 5.
2. Methods
2.1. First-principles calculations
In the present calculations, the orthorhombic supercells of the HCP, SFs and LPSOs structures of Mg and Mg alloys are utilized, with an orientation relationship between the primitive hcp Mg and the supercell of ,
and
(more details can be found in our previous work [Citation10,Citation15,Citation31]). The optimized structures and energies of the investigated structures are obtained by using the Vienna ab initio simulation package [Citation32,Citation33] with the generalized gradient approximation [Citation34] for the exchange-correction functional and the projector augmented wave [Citation35] for the electron–ion interaction. The plane wave cutoff energy is set as 387 eV, that is, 1.4 times the default cutoff energy for high-accuracy calculations, and the energy convergence criterion of electronic self-consistency is 10−6 eV/atom. At the ground state, the wave functions are sampled on γ -centered Monkhorst-Pack grids generated automatically with the same scaling length l = 60. While the structures are fully relaxed by the Methfessel-Paxton technique [Citation36], the final total energy calculations and the eDOS calculations are performed by the tetrahedron method incorporating Blöchl correction [Citation37]. Bonding charge density (Δρ) [Citation38–41] is used to characterize the electronic structures of the investigated structures. It is calculated through the charge density difference between the fully relaxed structure of self-consistent calculations and the same structure of non self-consistent calculations.
2.2. Phonon calculations
The supercell approach via the Yphon package [Citation42–44] together with Vienna ab initio simulation package [Citation32,Citation33] is used to predict the phonon frequencies of SFs and LPSOs in Mg. The capability of Yphon package yielding accurate phonon frequencies has been shown in the application of polar materials [Citation42] and random alloys [Citation44] since all of the interaction force constants between the atom within the supercell are included [Citation45]. The phonon frequencies can be calculated by solving the eigenvalue problems of the reciprocal dynamic matrix , shown as follows:
(1)
where α and β are the Cartesian axes of either x, y or z;
the wave vector; l the phonon mode and
the corresponding normalized atomic displacement weighted by the square root of the atomic mass. Within the supercell approach, the dynamic matrix
is defined from the primitive unit cell of the ideal lattice, through the following Fourier transformation [Citation42]:
(2)
where uj is atomic mass of the jth atom in the primitive cell of the ideal lattice, P the index of the primitive unit cell of the ideal lattice in the supercell,
is the cumulative force constant between the atom positioned at R(P, j) and the atom positioned at R(P, k). R(P) the position of the Pth primitive unit cell in the phonon supercell. N is the supercell size in terms of the number of primitive unit cell of the ideal lattice. The procedure to calculate the phonon properties of Mg with SFs and LPSOs is the same as that used in the calculation of random alloy [Citation44].
Based on the predicted LPDOS, the lattice vibrational contribution to Helmholtz energy and vibrational entropy can be calculated through [Citation46–50]
(3)
(4)
where
is the Boltzmann constant; T the temperature and
the phonon DOS as a function of phonon frequency ω at volume V.
3. Results
3.1. Phonon and thermodynamic properties of Mg
The phonon dispersion curve of Mg at the equilibrium volume compared with the available experimental data [Citation51,Citation52] is shown in Figure (a). A good agreement between the current theoretical calculation and the inelastic-neutron-scattering measurement indicates the accurate calculations of the force constants and the reliability of the current approach. Thus, the same parameters in the first-principles calculations are applied in the study of LPDOS of the atoms occupying positions in fault layers.
Figure 1. Phonon dispersion curve and phonon density of state (DOS) of Mg at the equilibrium volume along with the inelastic-neutron-scattering measurement at 290 K (labeled as Mg-Ref) [Citation51,Citation52], (a) HCP Mg; (b) stacking faults (growth fault—I1; deformation fault—I2; and extrinsic faults—EF)and (c) LPSOs (6H, 10H, 14H, 18R, 24R).
![Figure 1. Phonon dispersion curve and phonon density of state (DOS) of Mg at the equilibrium volume along with the inelastic-neutron-scattering measurement at 290 K (labeled as Mg-Ref) [Citation51,Citation52], (a) HCP Mg; (b) stacking faults (growth fault—I1; deformation fault—I2; and extrinsic faults—EF)and (c) LPSOs (6H, 10H, 14H, 18R, 24R).](/cms/asset/bbdcfa7c-229f-4ae7-a526-f0e9cabbf208/tmrl_a_1308973_f0001_c.jpg)
Comparing with the total phonon DOS of Mg, it can be seen that the frequency of the phonon peaks (ω > 7 THz) of SFs has a shift toward a high-frequency mode as shown in Figure (b). Similarly, the displacement feature has been captured when investigating the LPDOS of Σ5(310) grain boundaries in Al (ωmax increases from 10 THz of the bulk to 11 THz of the boundary) [Citation27]. Since there is no the coincident site lattice at the boundaries (the proposed type-2 grain boundary in [Citation27]), this displacement toward the high-frequency region could be caused by the tensile strain around the fault layers. It can be seen that there is no change in the peak position of low-frequency mode (ω < 4 THz) for growth fault (I1) and extrinsic fault (EF), compared to that of perfect Mg. Moreover, no significant change in width is observed. On the contrary, the positions of the phonon peaks (ω > 7 THz) of SFs, including the deformation fault (I2), shift toward the high-frequency mode.
Figure (c) shows the contributions of fault layers to the phonon DOS of Mg with various LPSOs in comparison with the available experimental data. It can be seen that the frequency of the phonon peaks (ω > 6.5 THz) of LPSOs shifts to the right part with a high-frequency mode. Compared to the LPDOS of perfect Mg, there is no change in the low-frequency mode (ω < 3.5 THz) for all the LPSOs. However, the significant change of the first peak of LPDOS (ω = 4 THz) of Mg with LPSOs is observed, whose height and width shrink in the order of 10H > 18R > 14H > 6H > 24H. Additionally, there is no peak at ω = 4 THz in 24H. As for the high-frequency mode (ω > 6.5 THz), no obvious change of the height and width occurs in 10H LPSO, compared with that of perfect Mg. However, the height of the second peak of LPDOS of Mg with LPSOs decreased in the order of 6H > 14H > 18R > 24R and the position of the second peak moves to the higher frequency mode in the same order. This is because the bond length and the stretching force constant corresponding to the first nearest neighbor change significantly with the formation of fault layers in the HCP lattice (more information can be found in Figure S2). Therefore, it can be concluded that the displacement of the phonon DOS toward the high-frequency mode of SFs is caused by the reduced bond length while the change of low-frequency mode is due to the elongated bond length. More interestingly, the reduction of stretching force constant at larger bond length of the first nearest neighbor indicates the increase in vibrational entropy.
To investigate the lattice stability of SFs and LPSOs of Mg alloys, the contribution of the free energy of harmonic and anharmonic effect on lattice vibration is essential to be considered. Within the quasiharmonic approximation, the anharmonic effect is accounted by the harmonic approximation at several volumes [Citation29,Citation53]. In this case, the Helmholtz energy can be approximated as
(5)
where E0 is the first-principles ground state energy at 0 K and volume V,
is the phonon contributions to the free energy given by Equation (3) at each V and
the electronic contributions with respect to the corresponding V and T. In this work, the first-principles calculations are carried out at P = 0 GPa, and hence PV = 0. Consequently, the Helmholtz energy is equal to the Gibbs energy. Moreover, compared to the
,
plays the dominant role in stabilizing the materials [Citation29,Citation53,Citation54]. Therefore, based on the variations of local PDOS, the key factor—
is only discussed in the following and the lattice vibrations caused by the fault layers of SFs and LPSOs provide the guidance to stabilize the structures.
Figure displays the contributions of fault layers to the vibrational entropy and Helmholtz energy (Fvib) along with their bond structures characterized by the deformation electron density isosurface. As shown in Figure (a), the atoms occupying the first nearest neighbor layer of a fault layer, such as L1, L3, L5, and L7, play an important role in yielding the excess frequency mode at ω = 7.25 THz. The values of phonon DOS of fault layers (L4 and L8) in the low-frequency and middle-frequency regions are larger than that of the non-fault layer. Moreover, LPDOS of the first nearest neighbor layers of the fault layers (L3 and L7) shows a shift toward the low-frequency region. Vibrational contributions to Helmholtz energy and entropy at constant volume of each atomic layer in growth fault are shown in Figure (a). It can be seen that the fault layers (L8) together with its near neighbor could stabilize the growth fault at high temperature since their entropy is higher, consistent with the displacement of LPDOS toward the low-frequency region.
Figure 2. Contributions of fault layers to the vibrational entropy and Helmholtz energy (Fvib) referring to their bond structures (Δρ = 0.0021 e−Å3 isosurface), (a) growth fault—I1; (b) deformation fault—I2; and (c) extrinsic fault—EF.
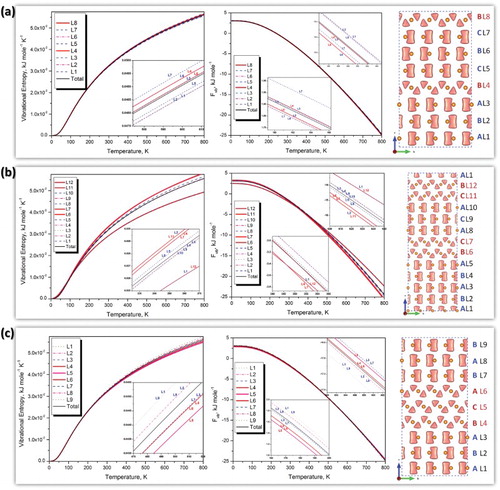
Similarly, LPDOS of the atoms in each layer of I2 and EF together with their bond structure characterized by the 0.5Δρmax charge density isosurface are presented in Figure (b,c), respectively. It can be seen that the low-frequency mode of atoms in fault layers (L6, L7, L11 and L12 in I2; L4, L5, and L6 in EF) are dominant. For example, the LPDOS peaks of L6 in I2 and L4 in EF at ω = 3.75 THz are higher than those in the non-fault layers (more information can be found in Figures S3 and S4). On the contrary, their peaks at ω = 7.5 THz are lower than those in the non-fault layers. It is necessary to point out that the LPDOS of L5 in EF is different than those of L4 and L6 at ω = 4.5 THz. This is caused by the difference of their interactions with the first nearest neighbor, matching well with their bond structures shown in Figure (b). Correspondingly, fault layers (L6, L7, and L11) could stabilize the deformation fault at high temperature since their entropy is higher. However, non-fault layers far away from the fault layer (L1 and L9) in EF seem to play a significant role to make the fault structure stable at high temperature since their LPDOS values at high frequency are dominant and larger than fault layers, which is caused by the shorter bond length or compressive strain in these atomic layers. Importantly, the changes in the PDOS provide the phonon contribution to the transition entropy [Citation54]. Our observation is in line with the result obtained by Iikubo et al., that the change in the atomic bonding state plays a significant role in the structural phase transition between 2H-18R and 2H-14H [Citation30].
3.2. Effect of solutes on the dislocation width of Mg-X
Based on our previous work presenting the contributions of solute atoms to stacking fault energy, segregation energy, shear modulus, bulk modulus, ideal shear stress, and bond strength [Citation31,Citation55,Citation56], the impact of solute atoms on the partial dislocation width of Mg alloys is discussed. It is commonly known that high strength and ductility will be obtained by reducing the partial dislocation width thus enhancing the density of the dislocation [Citation57–59]. In the basal plane, the dissociation of a perfect dislocation into two Shockley partial dislocations (i.e. ) would result in the formation of SFs [Citation55,Citation60]. The distance between these two Shockley partial dislocations, d, depends on the stacking fault energy (γ) and can be expressed as [Citation60]
(6)
where G is the shear modulus, b the magnitude of the Burgers vector of the Shockley partial dislocations,
the angle between two Shockley partial dislocations, and
the Poisson ratio.
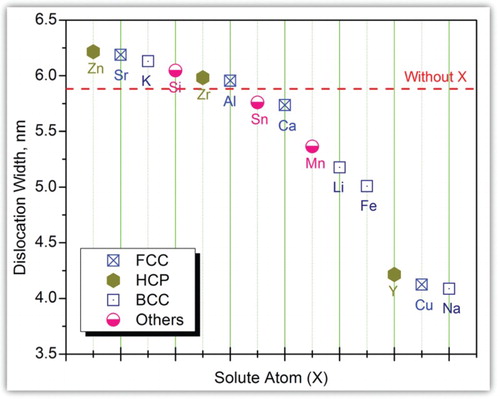
Therefore, through combining effects of solute atoms on the γ of I2 [Citation31], the G and (Tables S2–S4 in the supplementary data), the predicted partial dislocation width of Mg-X is decreased in the order of Zn > Sr > K > Si > Zr > Al > Sn > Ca > Mn >Li > Fe > Y > Cu > Na, shown in Figure . It is worth to highlight that Na cannot be the best alloying element to strengthen Mg alloys although it reduces the d to the smallest value. This is because the bonding strength of the interface will be weakened by the segregation of the Na [Citation31,Citation61,Citation62]. Similarly, bonding strength of the interface will be decreased by the segregation of the Cu [Citation55]. Herein, Y should be the primary alloying element among those investigated ones to strengthen Mg alloys. Recently, it has been reported that the presence of a supersaturated solid solution strengthening for basal slip and the enhanced activity of prismatic slip are the major reasons for the anomaly mechanical behavior of Mg-Y alloys [Citation63,Citation64].
Figure 3. Partial dislocation width of in Mg-X alloys affected by solute atoms (X). Various symbols are used to identify the crystal structures of each individual solute atom at room temperature, such as FCC, BCC, HCP, and others.
3.3. Case study of bonding structure and strength in Mg-Gd-TM (TM = Zn and Zr)
In this section, bond structure and strength of Mg97.5Gd1.67TM0.83 alloys affected by the solute atoms (Zn and Zr) and the fault layers in 6H and 10H LPSOs are discussed in terms of the bonding charge density (Δρ) and eDOS. Figure shows the isosurfaces of the bonding charge density (Δρ = 0.0021 e−Å3) of atomic cluster of Gd-TM in 6H and 10H LPSOs of Mg97.5Gd1.67TM0.83 (TM = Zn and Zr). It can be seen that the Δρ of atomic cluster of Gd-TM is significantly higher than that of surrounding Mg atoms, whose bonding charge densities are further decreased due to the alloying elements. This is because (i) more electrons locate at the position of atomic clusters of alloying elements since they have more valence electrons, for example, according to the electronic configurations of solute atoms in the first-principles calculations, 12 electrons for the valance of Zn (3d104s2) and Zr (4s24p64d25s2); and (ii) contributions of solute atoms and fault layers to the redisctribution of electrons result in the electron density change. The enhanced electrons along the basal plane caused by atomic cluster of Gd-TM (the gray rectangles highlighted in Figure ) and the reduced electrons in the prismatic and pyramidal planes (the yellow and blue rectangles highlighted in Figure ) indicate the bonds are strengthened along basal plane but weakened along prismatic and pyramidal planes. When the solute atoms of Gd form an atomic array with TM locating the first nearest neighbor site, the bond morphology will be significantly changed, compared with that of atomic cluster of Gd-TM. Moreover, the bond strength along prismatic planes dramatically decreased by the formation of atomic array in 6H of Mg97.5Gd1.67Zn0.83 and Mg97.5Gd1.67Zr0.83. Furthermore, the basal plane of Mg is strengthened due to the formation of a stronger chemical bond between atomic array/cluster and the Mg matrix, revealing the strengthening mechanism of RE and TM to Mg-TM-RE alloys (more information can be found in Figure S6 and [Citation65]). On the contrary, the weakened bond strength of the Mg matrix in the prismatic plane by the fault layers and alloying elements indicates a possible non-basal slip systems could occur during deformation, which could improve the ductility of Mg alloys. The directional bond will result in the elastic anisotropy, and thus hinder the anisotropic of deformation behavior (more information about the correlation between bonding and elastic anisotropy can be found in [Citation15,Citation38]).
Figure 4. Prismatic plane ((100)S.C.) view of Δρ = 0.0021 e−/Å3 isosurface plots Mg97.5TM0.83Gd1.67 (TM = Zn and Zr) with atomic array of Gd and TM, (a) 6H and (b) 10H. The numbers 1 and 2 are applied to identify the inequivalent Gd atoms with and without Gd-TM bond in 10H, respectively. The process constructing the supercell of 10H LPSO can be found in [Citation65].
![Figure 4. Prismatic plane ((100)S.C.) view of Δρ = 0.0021 e−/Å3 isosurface plots Mg97.5TM0.83Gd1.67 (TM = Zn and Zr) with atomic array of Gd and TM, (a) 6H and (b) 10H. The numbers 1 and 2 are applied to identify the inequivalent Gd atoms with and without Gd-TM bond in 10H, respectively. The process constructing the supercell of 10H LPSO can be found in [Citation65].](/cms/asset/e7019642-506e-414b-a60a-e366798e9c60/tmrl_a_1308973_f0004_c.jpg)
In order to reveal the electronic origin of the interactions between solute atoms and Mg atoms, the total and partial eDOS for Mg97.5TM0.83Gd1.67 (TM = Zn and Zr) with 6H and 10H LPSOs are calculated and are shown in Figure . In particular, the partial eDOS curves clearly present the electronic hybridizations of valance electron states of the d-electrons of Zn and Zr atoms, the f-electron Gd atoms and the p-electron Mg atoms in 6H and 10H LPSOs. In line with the bonding structure characterizations shown in Figure , the positive values of eDOS at the Fermi level indicate the metallic behavior of those structures. Attributed to the segregations of the solute atoms at the fault layers of 6H LPSO, the Zn-p states, Zr-p states, Gd-f states, Mg-s, and Mg-p states are very strong both below and above the Fermi level, indicating their strong hybridization, see Figure (a,b). While in the 10H LPSO, the valence states of Zn, Zr, Gd, and Mg play an almost identical contribution below the Fermi level. On the contrary, at the Fermi level of 10H, the Mg-p states, Zn-p states, Zr-p states, and Gd-p states are generally higher than the corresponding Mg-s states, Zn-d states, Zr-d states, and Gd-f states, respectively, see Figure (c,d).
Figure 5. The total and partial eDOS for Mg97.5TM0.83Gd1.67 (TM = Zn and Zr), (a) and (b) 6H LPSO; (c) and (d) 10H LPSO. The blue dotted lines at 0 eV dictate the Fermi level.
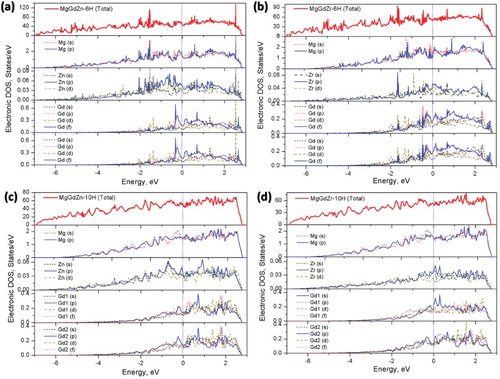
Moreover, from the common features of the partial eDOS of 6H and 10H LPSOs of Mg97.5TM0.83Gd1.67, we can see that the Fermi levels of Mg(s and p) and Zn (s, p and d) locate at the local maximum, while those of Gd (s, p, d and f) locate at the local minimum. The Fermi levels of Zr (s, p and d) locate at a local minimum of the partial eDOS of 10H LPSO and at a local maximum of the partial eDOS of 6H LPSO. Since a reduction in eDOS at the Fermi level will contribute to stabilize the given structure [Citation66,Citation67], the hybridizations of Mg-s states, Zn-d states, Zr-d states, and Gd-f states would benefit not only to the stability of the 10H LSPO but also to the strong bonding, thus resulting in an improvement of the strength of Mg97.5TM0.83Gd1.67. This conclusion is in line with the aforementioned free energy analysis of the SFs and LPSOs at high temperatures in Figure and the bond strength characterizations of LPSOs in Figure . Furthermore, according to the general features of the eDOS of 6H and 10H LPSOs of Mg97.5TM0.83Gd1.67, the so-called quasi-gap near the Fermi level of the total eDOS implies the presence of strong covalent bonding [Citation67], which increases the strength of the materials. This feature agrees well with the bonding charge density analysis discussed above, which is also similar to the observations in 6H LPSO of Mg94Y1Zn1 [Citation67]. Thus, through integrating the characterizations of Δρ and eDOS, the atomic and the electronic basis for the solid solute strengthening mechanism is revealed by the electronic redistributions, providing a fundamental insight into the ‘strengthening’ of the Mg alloys.
4. Discussions
In order to strengthen Mg alloys, two strategies are applied in the present work, which are the modification of the matrix geometry by planar faults and the optimization the local lattice strain by solute atoms. Accordingly, the deformation anisotropy could be reduced by enhancing the bonding strength of the basal plane but weakening that of the prismatic and the pyramidal planes, thus improving the strength and the ductility of Mg alloys. This is because (i) the basal slip of Mg and Mg alloys is very easy to trigger due to the extremely high plastic anisotropy (the ratio of the critical resolved sheer stress between basal slip and non-basal slip is about 102) [Citation68] and (ii) without non-basal slip or twinning, the basal slip cannot accommodate arbitrary shape change and excessive basal slip in combination with unrelaxed tensile stresses in the plane-normal direction leading to spatially localized damage and rapid fracture [Citation5].
Based on the deformation electron density isosurface, the directional bond of Mg matrix will result in the elastic anisotropy, and thus hinders the anisotropic deformation behavior. With the formation of fault layers in the matrix of Mg, the rod-type directional bonds transfer into tetrahedrons, which are the typical FCC-type chemical bonds [Citation38]. Moreover, the inhomogeneous electron distribution in the fault layers of SFs and LPSOs could be introduced and enhanced by the interactions among solute atoms and fault layers. Since it is more difficult for the electrons to readapt during breaking rod-type directional bonds than the spherical ones [Citation69], the redistribution of electrons characterized by the change of bond morphology implies the directionality of the bonds crossing the fault layers of LPSOs and the dependency of formation energy of defects on composition [Citation70]. Furthermore, the fault layers of LPSOs enriched of solute atoms [Citation13,Citation71,Citation72] have a classical nano-lamellar feature (3–6 non-fault layers of the Mg matrix are separated by 1 or 2 fault layers [Citation10]), promising an enhancement of pinning effect during deformation. Due to the segregation of the clusters of solute atoms to the fault layers, both the interlayer bonding strength could be improved, resulting in the enhancement of interface shear stress (ISS, defined as the shear stress along interface [Citation73]) of the basal plane thus reducing the plastic anisotropy and promoting the plastic flow of the nano-lamellar structure. It has been observed that the nano-lamellar metals can absorb dislocations into the interface through dislocation-interface reactions, suggesting a new mechanism on shear stress-driven interface-mediated plasticity in the confined nano-lamellar structures [Citation73].
5. Summary
We present the strategies to strengthen Mg alloys through modifying the matrix geometry by planar faults and the local lattice strain by solute atoms. It is found that the anomalous shifts of the LPDOS of SFs and LPSOs toward the high-frequency mode relate to the HCP-FCC bond transformation, resulting in the increase of vibrational entropy and the decrease of free energy to stabilize the corresponding structures. It is understood that due to the formation of the fault layers and the segregation of the clusters of solute atoms to fault layers, the deformation anisotropy could be reduced by enhancing the bonding strength of the basal plane but weakening that of the prismatic and the pyramidal planes, thus improving the strength and the ductility of Mg alloys. The partial eDOS curves clearly reveal the electronic hybridizations of valance electron states of the d-electron Zn and Zr atoms, the f-electron Gd atoms and the p-electron Mg atoms in 6H and 10H LPSOs. According to the common features of the eDOS of 6H and 10H LPSOs of Mg97.5TM0.83Gd1.67, the quasi-gap near the Fermi level of the total eDOS implies the presence of strong covalent bonding and indicates the improvement of the strength of the materials. Through integrating the characterizations of bonding charge density and eDOS, the atomic and the electronic basis for the solid solute strengthening mechanism is revealed by the electron redistributions, providing a fundamental insight into the ‘strengthening’ of Mg alloys.
Supplementary_Material
Download MS Word (6.4 MB)Acknowledgements
First-principles calculations were carried out on the LION clusters at the Pennsylvania State University supported by the Materials Simulation Center and the Research Computing and Cyberinfrastructure unit at the Pennsylvania State University. Calculations were also carried out on the CyberStar.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
William Yi Wang http://orcid.org/0000-0002-8814-525X
Shun Li Shang http://orcid.org/0000-0002-6524-8897
Laszlo J. Kecskes http://orcid.org/0000-0002-1342-3729
Zi-Kui Liu http://orcid.org/0000-0003-3346-3696
Additional information
Funding
Related Research Data
References
- Shang S, Zhang H, Ganeshan S, et al. The development and application of a thermodynamic database for magnesium alloys. Jom. 2008;60:45–47. doi: 10.1007/s11837-008-0165-1
- Pollock TM. Weight loss with magnesium alloys. Science. 2010;328:986–987. doi: 10.1126/science.1182848
- Chen Q, Thouas GA. Metallic implant biomaterials. Mater Sci Eng R-Rep. 2015;87:1–57. doi: 10.1016/j.mser.2014.10.001
- Chen L-Y, Xu J-Q, Choi H, et al. Processing and properties of magnesium containing a dense uniform dispersion of nanoparticles. Nature. 2015;528:539–543. doi: 10.1038/nature16445
- Yu Q, Qi L, Mishra RK, et al. Reducing deformation anisotropy to achieve ultrahigh strength and ductility in Mg at the nanoscale. Proc Natl Acad Sci USA. 2013;110:13289–13293. doi: 10.1073/pnas.1306371110
- Agnew SR, Duygulu O. Plastic anisotropy and the role of non-basal slip in magnesium alloy AZ31B. Int J Plast. 2005;21:1161–1193. doi: 10.1016/j.ijplas.2004.05.018
- Yoo MH. Slip, twinning, and fracture in hexagonal close-packed metals. Metall Trans A. 1981;12:409–418. doi: 10.1007/BF02648537
- Nie JF, Zhu YM, Liu JZ, et al. Periodic segregation of solute atoms in fully coherent twin boundaries. Science. 2013;340:957–960. doi: 10.1126/science.1229369
- Egusa D, Abe E. The structure of long period stacking/order Mg-Zn-RE phases with extended non- stoichiometry ranges. Acta Mater. 2012;60:166–178. doi: 10.1016/j.actamat.2011.09.030
- Wang WY, Shang SL, Wang Y, et al. Electronic structures of long periodic stacking order structures in Mg: a first-principles study. J Alloy Compd. 2014;586:656–662. doi: 10.1016/j.jallcom.2013.10.068
- Clouet E, Lae L, Epicier T, et al. Complex precipitation pathways in multicomponent alloys. Nat Mater. 2006;5:482–488. doi: 10.1038/nmat1652
- Inoue A, Matsushita M, Kawamura Y, et al. Novel hexagonal structure of ultra-high strength magnesium-based alloys. Mater Trans. 2002;43:580–584. doi: 10.2320/matertrans.43.580
- Hui X, Dong W, Chen GL, et al. Formation, microstructure and properties of long-period order structure reinforced Mg-based bulk metallic glass composites. Acta Mater. 2007;55:907–920. doi: 10.1016/j.actamat.2006.09.012
- Jian WW, Cheng GM, Xu WZ, et al. Ultrastrong Mg alloy via nano-spaced stacking faults. Mater Res Lett. 2013;1:61–66. doi: 10.1080/21663831.2013.765927
- Wang WY, Shang SL, Wang Y, et al. Electron localization morphology of the stacking faults in Mg: a first-principles study. Chem Phys Lett. 2012;551:121–125. doi: 10.1016/j.cplett.2012.09.028
- Argon AS, Moffatt WC. Climb of extended edge dislocations. Acta Metall. 1981;29:293–299. doi: 10.1016/0001-6160(81)90156-5
- Chetty N, Weinert M. Stacking faults in magnesium. Phys Rev B. 1997;56:10844–10851. doi: 10.1103/PhysRevB.56.10844
- Lu K, Lu L, Suresh S. Strengthening materials by engineering coherent internal boundaries at the nanoscale. Science. 2009;324:349–352. doi: 10.1126/science.1159610
- Zhao YH, Zhu YT, Liao XZ, et al. Tailoring stacking fault energy for high ductility and high strength in ultrafine grained Cu and its alloy. Appl Phys Lett. 2006;89:121906. doi: 10.1063/1.2356310
- Jian WW, Cheng GM, Xu WZ, et al. Physics and model of strengthening by parallel stacking faults. Appl Phys Lett. 2013;103:133108. doi: 10.1063/1.4822323
- Zheng B, Ertorer O, Li Y, et al. High strength, nano-structured Mg-Al-Zn alloy. Mater Sci Eng A. 2011;528:2180–2191. doi: 10.1016/j.msea.2010.11.073
- Li Y, Kong QP. On the relationship between creep rate and stacking-fault energy. Phys Status Solidi A. 1989;113:345–351. doi: 10.1002/pssa.2211130212
- Chookajorn T, Murdoch HA, Schuh CA. Design of stable nanocrystalline alloys. Science. 2012;337:951–954. doi: 10.1126/science.1224737
- van de Walle A, Ceder G. The effect of lattice vibrations on substitutional alloy thermodynamics. Rev Mod Phys. 2002;74:11–45. doi: 10.1103/RevModPhys.74.11
- Pomrehn GS, Toberer ES, Snyder GJ, et al. Entropic stabilization and retrograde solubility in Zn4Sb3. Phys Rev B. 2011;83:094106. doi: 10.1103/PhysRevB.83.094106
- Shang S-L, Wang Y, Du Y, et al. Entropy favored ordering: phase stability of Ni3Pt revisited by first-principles. Intermetallics. 2010;18:961–964. doi: 10.1016/j.intermet.2010.01.011
- Hashimoto M, Ishida Y, Yamamoto R, et al. Computer-simulation of the structure and atomic vibration of the sigma 5 tilt boundary in aluminum. J Phys F: Metal Phys. 1980;10:1109–1116. doi: 10.1088/0305-4608/10/6/011
- Creuze J, Berthier F, Tetot R, et al. Intergranular segregation and vibrational effects: a local analysis. Phys Rev B. 2000;61:14470–14480. doi: 10.1103/PhysRevB.61.14470
- Zhang H, Shang SL, Wang Y, et al. First-principles calculations of the elastic, phonon and thermodynamic properties of Al12Mg17. Acta Mater. 2010;58:4012–4018. doi: 10.1016/j.actamat.2010.03.020
- Iikubo S, Matsuda K, Ohtani H. Phase stability of long-period stacking structures in Mg-Y-Zn: a first-principles study. Phys Rev B. 2012;86:054105. doi: 10.1103/PhysRevB.86.054105
- Wang WY, Shang SL, Wang Y, et al. Effects of alloying elements on stacking faults energies and electronic structures of binary Mg alloys: a first-principles study. Mater Res Lett. 2014;2:29–36. doi: 10.1080/21663831.2013.858085
- Kresse G, Furthmuller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B. 1996;54:11169–11186. doi: 10.1103/PhysRevB.54.11169
- Kresse G, Furthmuller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp Mater Sci. 1996;6:15–50. doi: 10.1016/0927-0256(96)00008-0
- Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B. 1999;59:1758–1775. doi: 10.1103/PhysRevB.59.1758
- Wang Y, Perdew JP. Correlation hole of the spin-polarized electron gas, with exact small-wave-vector and high-density scaling. Phys Rev B. 1991;44:13298–13307. doi: 10.1103/PhysRevB.44.13298
- Methfessel M, Paxton AT. High-precision sampling for Brillouin-zone integration in metals. Phys Rev B. 1989;40:3616–3621. doi: 10.1103/PhysRevB.40.3616
- Blochl PE, Jepsen O, Andersen OK. Improved tetrahedron method for Brillouin-zone integrations. Phys Rev B. 1994;49:16223–16233. doi: 10.1103/PhysRevB.49.16223
- Nakashima PNH, Smith AE, Etheridge J, et al. The bonding electron density in aluminum. Science. 2011;331:1583–1586. doi: 10.1126/science.1198543
- Midgley PA. Electronic bonding revealed by electron diffraction. Science. 2011;331:1528–1529. doi: 10.1126/science.1203614
- Wang WY, Shang SL, Wang Y, et al. Lattice distortion induced anomalous ferromagnetism and electronic structure in FCC Fe and Fe-TM (TM = Cr, Ni, Ta and Zr) alloys. Mater Chem Phys. 2015;162:748–756. doi: 10.1016/j.matchemphys.2015.06.051
- Wang WY, Darling KA, Wang Y, et al. Power law scaled hardness of Mn strengthened nanocrystalline AlMn non-equilibrium solid solutions. Scripta Mater. 2016;120:31–36. doi: 10.1016/j.scriptamat.2016.04.003
- Wang Y, Wang JJ, Wang WY, et al. A mixed-space approach to first-principles calculations of phonon frequencies for polar materials. J Phys: Condes Matter. 2010;22:202201.
- Wang Y, Wang JJ, Saal JE, et al. Phonon dispersion in Sr2RuO4 studied by a first-principles cumulative force-constant approach. Phys Rev B. 2010;82:172503. doi: 10.1103/PhysRevB.82.172503
- Wang Y, Zacherl CL, Shang SL, et al. Phonon dispersions in random alloys: a method based on special quasi-random structure force constants. J Phys: Condes Matter. 2011;23:485403.
- Parlinski K, Li ZQ, Kawazoe Y. First-principles determination of the soft mode in cubic ZrO2. Phys Rev Lett. 1997;78:4063–4066. doi: 10.1103/PhysRevLett.78.4063
- Shang SL, Wang J, Wang Y, et al. Phonon and thermodynamic properties of Al-Mn compounds: a first-principles study. Comp Mater Sci. 2011;50:2096–2103. doi: 10.1016/j.commatsci.2011.02.015
- Shang SL, Wang WY, Wang Y, et al. Temperature-dependent ideal strength and stacking fault energy of fcc Ni: a first-principles study of shear deformation. J Phys: Condes Matter. 2012;24:155402.
- Wrobel J, Hector Jr LG, Wolf W, et al. Thermodynamic and mechanical properties of lanthanum-magnesium phases from density functional theory. J Alloy Compd. 2012;512:296–310. doi: 10.1016/j.jallcom.2011.09.085
- Zhang H, Shang S-L, Wang Y, et al. Thermodynamic properties of Laves phases in the Mg–Al–Ca system at finite temperature from first-principles. Intermetallics. 2012;22:17–23. doi: 10.1016/j.intermet.2011.08.019
- Shang SL, Wang Y, Kim D, et al. First-principles thermodynamics from phonon and Debye model: application to Ni and Ni3Al. Comp Mater Sci. 2010;47:1040–1048. doi: 10.1016/j.commatsci.2009.12.006
- Althoff JD, Allen PB, Wentzcovitch RM, et al. Phase-diagram and thermodynamic properties of solid magnesium in the quasi-harmonic approximation. Phys Rev B. 1993;48:13253–13260. doi: 10.1103/PhysRevB.48.13253
- Pynn R, Squires GL. Measurements of the normal-mode frequencies of magnesium. Proc Roy Soc Lond A. 1972;326:347–360. doi: 10.1098/rspa.1972.0013
- Shang SL, Wang Y, Liu ZK. First-principles calculations of phonon and thermodynamic properties in the boron-alkaline earth metal binary systems: B-Ca, B-Sr, and B-Ba. Phys Rev B. 2007;75:024302. doi: 10.1103/PhysRevB.75.024302
- Budai JD, Hong J, Manley ME, et al. Metallization of vanadium dioxide driven by large phonon entropy. Nature. 2014;515:535–539. doi: 10.1038/nature13865
- Shang SL, Wang WY, Zhou BC, et al. Generalized stacking fault energy, ideal strength and twinnability of dilute Mg-based alloys: A first-principles study of shear deformation. Acta Mater. 2014;67:168–180. doi: 10.1016/j.actamat.2013.12.019
- Ganeshan S, Shang SL, Wang Y, et al. Effect of alloying elements on the elastic properties of Mg from first-principles calculations. Acta Mater. 2009;57:3876–3884. doi: 10.1016/j.actamat.2009.04.038
- Chen B, Lutker K, Raju SV, et al. Texture of nanocrystalline nickel: probing the lower size limit of dislocation activity. Science. 2012;338:1448–1451. doi: 10.1126/science.1228211
- Wu Z, Curtin WA. Intrinsic structural transitions of the pyramidal I<c + a> dislocation in magnesium. Scripta Mater. 2016;116:104–107. doi: 10.1016/j.scriptamat.2016.01.041
- Ghazisaeidi M, Hector Jr LG, Curtin WA. First-principles core structures of edge and screw dislocations in Mg. Scripta Mater. 2014;75:42–45. doi: 10.1016/j.scriptamat.2013.11.013
- Zhu YM, Morton AJ, Weyland M, et al. Characterization of planar features in Mg-Y-Zn alloys. Acta Mater. 2010;58:464–475. doi: 10.1016/j.actamat.2009.09.025
- Lu G-H, Zhang Y, Deng S, et al. Origin of intergranular embrittlement of Al alloys induced by Na and Ca segregation: grain boundary weakening. Phys Rev B. 2006;73:224115. doi: 10.1103/PhysRevB.73.224115
- Zhang S, Han Q, Liu ZK. Fundamental understanding of Na-induced high temperature embrittlement in Al-Mg alloys. Philos Mag. 2007;87:147–157. doi: 10.1080/14786430600941587
- Zhang D, Wen H, Kumar MA, et al. Yield symmetry and reduced strength differential in Mg-2.5Y alloy. Acta Mater. 2016;120:75–85. doi: 10.1016/j.actamat.2016.08.037
- Zhang D, Zheng B, Zhou Y, et al. Prism stacking faults observed contiguous to a {10–12} twin in a Mg-Y alloy. Scripta Mater. 2014;76:61–64. doi: 10.1016/j.scriptamat.2013.12.015
- Wang WY, Shang SL, Wang Y, et al. Solid-solution hardening in Mg-Gd-TM (TM = Ag, Zn, and Zr) alloys: an integrated density functional theory and electron work function study. JOM. 2015;67:2433–2441. doi: 10.1007/s11837-015-1555-9
- Fu CL, Wang XD, Ye YY, et al. Phase stability, bonding mechanism, and elastic constants of Mo5Si3 by first-principles calculation. Intermetallics. 1999;7:179–184. doi: 10.1016/S0966-9795(98)00018-1
- Chen P, Li DL, Yi JX, et al. Microstructure and electronic characteristics of the 6H-type ABACAB LPSO structure in Mg97Zn1Y2 alloy. J Alloy Compd. 2009;485:672–676. doi: 10.1016/j.jallcom.2009.06.045
- Hutchinson WB, Barnett MR. Effective values of critical resolved shear stress for slip in polycrystalline magnesium and other hcp metals. Scripta Mater. 2010;63:737–740. doi: 10.1016/j.scriptamat.2010.05.047
- Ogata S, Li J, Yip S. Ideal pure shear strength of aluminum and copper. Science. 2002;298:807–811. doi: 10.1126/science.1076652
- Qi Y, Mishra RK. Ab initio study of the effect of solute atoms on the stacking fault energy in aluminum. Phys Rev B. 2007;75:224105-1–224105-5. doi: 10.1103/PhysRevB.75.224105
- Abe E, Kawamura Y, Hayashi K, et al. Long-period ordered structure in a high-strength nanocrystalline Mg-1 at% Zn-2 at% Y alloy studied by atomic-resolution Z-contrast STEM. Acta Mater. 2002;50:3845–3857. doi: 10.1016/S1359-6454(02)00191-X
- Zhu YM, Morton AJ, Nie JF. The 18R and 14H long-period stacking ordered structures in Mg-Y-Zn alloys. Acta Mater. 2010;58:2936–2947. doi: 10.1016/j.actamat.2010.01.022
- Yan JW, Zhu XF, Yang B, et al. Shear stress-driven refreshing capability of plastic deformation in nanolayered metals. Phys Rev Lett. 2013;110:155502. doi: 10.1103/PhysRevLett.110.155502