
ABSTRACT
Density functional theory (DFT) was utilized to compute the gravimetric capacity, volumetric capacity, and the binding energy of hydrogen molecules in silicon clathrates with guest (A) atoms such as Ba, Na, and Li, and framework substitutional atoms (M) such as C, Al, and Cu. The DFT computations show that these Type I intermetallic clathrates can accommodate a large number of hydrogen molecules, equivalent to 10 wt.%, and such hydrogenated structures, Ax(H2)nMySi46−y, occur with only a modest increase in lattice volume and a binding energy within the desirable range of 0.1–0.6 eV/H2 for hydrogen storage at or near ambient temperature.
GRAPHICAL ABSTRACT
IMPACT STATEMENT
This paper identifies a number of Type I silicon clathrates that can accommodate large amounts of hydrogen molecules (10 wt.%) and may be suitable as hydrogen storage materials.
In recent years, there has been great interest in developing materials-based reversible hydrogen storage systems for transportation applications, such as fuel cell vehicles and hydrogen combustion engines, that exhibit equal or greater gravimetric and volumetric storage capacities as compared with compressed gas storage (75 MPa), though at much lower and safer pressures. For a fuel cell power system in a vehicle operating directly on hydrogen, the near-term system-level gravimetric and volumetric storage targets proposed for net useable hydrogen are 5.5 wt.% and 0.040 kg H2/L, respectively, which includes the stored hydrogen, the container, and all auxiliary hardware [Citation1]. These system targets imply that the capacities on a material basis alone would have to be at least twice as much − on the order of ≥10 wt.% and ≥0.080 kg H2/L.
In light of these storage targets, new hydrogen storage materials need to be developed whose storage capacities can be achieved within practical thermodynamic regimes relative to pressure, temperature, and heat release; preferably, near room temperature and modest pressure (<25 MPa). Kinetic constraints on the rate of hydrogen uptake and discharge in prospective materials must also be limited to allow practical fill times (≤40 s/kg H2) of the storage system and mass flow rate to the fuel cell (≥0.02 g/s kW). One particular approach is to engineer micro- or mesoporous materials that can adsorb hydrogen molecules in their cavities at low or ambient temperatures and release them at a higher temperature. Candidate materials that have been considered for hydrogen storage include metal hydrides, zeolites, porous carbons, and metal–organic frameworks (MOFs) via storage mechanisms such as interstitial and complex hydride formation, physisorption, and the controversial H2 spillover effects in catalyst-doped heterogeneous materials [Citation2,Citation3]. The current capacity of some of the candidate hydrogen materials as a function of sorption temperature and mechanism is illustrated in Figure [Citation2] together with the material storage target desired for vehicle applications.
Figure 1. The relative gravimetric performance of physisorption, spillover, and chemisorption (complex hydride) materials towards achieving the minimum material-based targets for on-board reversible fuel storage (from Miller et al. [Citation2]).
![Figure 1. The relative gravimetric performance of physisorption, spillover, and chemisorption (complex hydride) materials towards achieving the minimum material-based targets for on-board reversible fuel storage (from Miller et al. [Citation2]).](/cms/asset/391c90fa-29cf-40a7-a9ea-c781b6ac147d/tmrl_a_1396261_f0001_c.jpg)
One of the hydrogen storage materials that is close to meeting the performance targets is the MOFs [Citation2,Citation3], whose crystalline frameworks with open structures and internal cavities can potentially accommodate and store large amounts of molecular hydrogen. Since surface area is qualitatively proportional to hydrogen gas uptake, molecular-scale engineering of MOFs has been rigorously employed to design and synthesize novel storage materials from a vast selection of readily assembled building units. MOF chemistry has, therefore, enabled a successful strategy for developing highly porous physisorption materials of extraordinary surface areas with well-defined, ordered, pore spaces. As illustrated in Figure , they have been shown to selectively accommodate a high capacity of hydrogen molecules reversibly, though so far only at low temperatures (77 K) [Citation2]. In the case of porous materials for hydrogen physisorption, two strategies—optimization of binding interactions and engineering of optimized structural motifs—are both necessary to achieve the volumetric and gravimetric targets for on-board storage of hydrogen.
After an extensive study [Citation2], it is now apparent that binding interactions must fall within an optimum range in order to maximize reversible storage at or close to room temperature. They can neither be too high nor too low, and the optimum range defines the thermodynamic constraints of the sorbent system. At or near room temperature, the desirable range for the binding energy of hydrogen, regardless of modality, is between 0.1 and 0.6 eV/H2 (10 and 60 kJ/mol). This range was determined from an analysis based on entropic arguments that the reference entropy values for H2 are between 0.1 and 10 MPa, and temperature ranging −20 to 85°C. At −20°C, the enthalpy of adsorption (ΔH = TΔS) ranges ∼21 to 32 kJ/mol H2 and at 85°C, the enthalpy increases to 51–71 kJ/mol H2. On this basis, one can determine that the ideal binding affinity falls in the range between ∼20 and 70 kJ/mol H2, leading to a conservative upper range of 58 kJ/mol H2 (0.6 eV/H2). None of the candidate materials compiled in Figure [Citation2] exhibits the desired binding energy range at the ambient temperatures.
Another crystalline framework with open structure that has been considered for hydrogen storage applications is inorganic Type I clathrates. Both K- and Na-stabilized Type I silicon clathrates with H2 encapsulation were synthesized [Citation4,Citation5]. Thermogravimetric measurements indicated that hydrogen desorption occurred at elevated temperatures (∼400°C), which make these materials impractical for on-board storage in vehicle applications unless modification can be made to lower the binding energy and the desorption temperature.
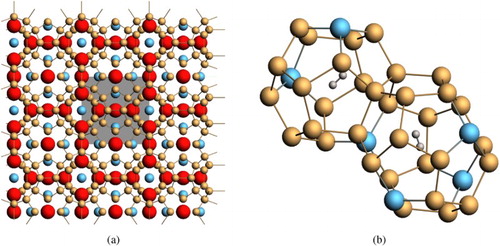
The structure of Type I intermetallic clathrates, shown in Figure , comprised a framework of X atoms forming a 3D cage structure. Type I silicon clathrate, Si46, consists of crystalline Si with a regular arrangement of 20-atom and 24-atom cages fused together through 5-atom pentagonal rings and 6-atom hexagonal rings. The crystal structure of the Si46 clathrate belongs to the space group [Citation6]. Some of the framework atoms can be substituted by atom M. The empty space within the cage structure can serve as host sites for guest atoms A. Type I clathrates can be described by the formula: AxMyX46−y [Citation6]. Representative framework, substitution, and guest atoms are listed in the caption of Figure . Recent investigations [Citation6,Citation7] have shown that both the guest atoms and the framework atoms on Type I silicon clathrates can be altered with other substitution atoms to create hybrid carbon and silicon clathrates that are metastable materials with tuneable physical and electronic properties by selective doping of guest and framework atoms. These developments provide a new avenue for investigating the possibility of novel metastable silicon-based clathrates for hydrogen storage applications.
Figure 2. (a) Extended crystal structure of the intermetallic clathrates (Type I). Red = volume space for guest atoms such as Ba, Na, and Li. Blue = substitutional framework atoms such as C, Al, and Cu. The unit cell is indicated by grey box. (b) Occupiable volume for H2 exists in the cages of both the 20-atom pentagonal dodecahedron clusters (2a Wyckoff sites) and 24-atom tetrakaidecahedron clusters (6d Wyckoff sites).
The objective of this article is to report the results of an investigation focused on identifying new Si-based clathrates that may be suitable for use as hydrogen storage materials in transportation applications. The approach is to apply first-principles computational methods based on density functional theory via the VASP code [Citation8] to compute the binding energy of hydrogen molecules in selected Type I Si-based clathrates. First-principle computational methods were used to design Si-based intermetallic clathrates with or without an alkali–metal guest atom, and Si framework atoms substituted with other elements such as C, Al, and Cu, to tune the void space for maximum H2 capacity and optimum binding interactions. The computational results indicate that the hydrogen binding energy of alloyed clathrates can be tailored to the optimum levels at or near ambient temperature desired for potential vehicle applications.
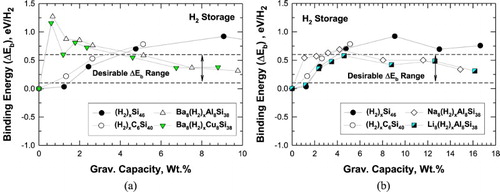
Figure (a) shows the density functional theory (DFT) results of the binding energy in eV/H2 for hydrogenation of Si46, C6Si40, Ba8Al8Si38, and Ba8Cu8Si38 as function of gravimetric capacity in weight percentage converted from the number of H2 molecules inserted in the cage structure. The desirable binding energy range for H2 at ambient temperature is also superimposed in Figure (a). The results indicate that the binding energy between hydrogen molecules and the interstitial cavities of Si46 and C6Si40 cage structures increases with the number of H2 molecules inserted or gravimetric capacity. In contrast, the hydrogen binding interactions for Ba8(H2)xAl8Si38 and Ba8(H2)xCu8Si38, which decrease within increasing gravimetric capacities, are within the desirable binding energy range at high gravimetric capacities. A similar comparison of the computed binding energy values with gravimetric capacity is presented in Figure (b) for hydrogenation Si46, C6Si40, Na8Al8Si38, and Li8Al8Si38. In this case, Figure (b) shows that Na8Al8Si38 and Li8Al8Si38 exhibit hydrogen binding interactions that are within the desirable range for the entire range of gravimetric capacity considered, ranging from 8 to 128 H2 molecules inserted. This finding suggests that Na8Al8Si38 and Li8Al8Si38 show high physisorption capacity for H2 with suitable binding energy values as potential hydrogen storage materials at or near ambient temperatures.
Figure 3. Predicted internal binding energy and capacity for intermetallic clathrates. The range over which binding interactions of H2 is favourable for room temperature storage is noted (a) hydrogenation of Si46, C6Si40, Ba8Al8Si38, and Ba8Cu8Si38 and (b) hydrogenation of Si46, C6Si40, Na8Al8Si38, and Li8Al8Si38.
The lattice parameters for individual unit cells of Si46, C6Si40, Ba8Al8Si30, and Ba8Cu8Si38 after various levels of hydrogenation are presented in Figure (a) as a function of gravimetric capacity. The lattice parameter for hydrogenated Ba8Al8Si38 and Ba8Cu8Si38 increase with increasing number of hydrogen molecules inserted (gravimetric capacity), while those of hydrogenated Si46 and C6Si40 do not exhibit similar increases with gravimetric capacity. Also superimposed in Figure (a) are the system-level storage targets for gravimetric and volumetric capacity prescribed by the U.S. Department of Energy (DOE) for on-board storage on vehicles for 2020 and the ultimate fleet targets [Citation1]. Recalling that, on a materials basis, the required gravimetric capacity would have to be approximately twice that of the system targets, the computational results nonetheless support the hypothesis that the hydrogenated Ba8Al8Si38 and Ba8Cu8Si38 clathrates can absorb large amounts of H2 molecules at or near room temperature, and may potentially achieve both the 2020 and ultimate system-level fleet targets prescribed by DOE, while empty Si46 and C6Si38 may not. A similar comparison of hydrogenated Si46, C6Si40, Na8Al8Si38, and Li8Al8Si38 is presented in Figure (b). Like Ba-stabilized silicon clathrate, the lattice parameters of Na- and Li-stabilized silicon clathrates increase with increasing number of hydrogen molecules inserted (gravimetric capacity), and also show promise toward achieving the system targets.
Figure 4. Predicted gravimetric capacity (materials basis) and lattice constant for intermetallic (hybrid) clathrates: (a) hydrogenation of Si46, C6Si40, Ba8Al8Si38 and Ba8Cu8Si38 and (b) hydrogenation of Si46, C6Si40, Na8Al8,Si38, and Li8Al8Si38. Vertical lines mark the system-level targets prescribed by DOE.
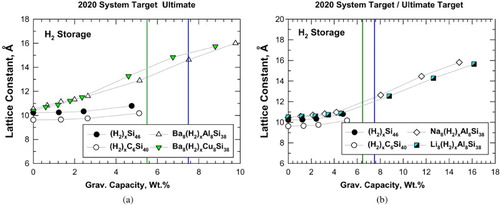
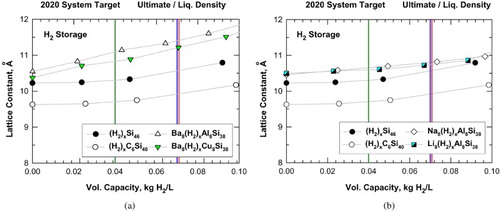
Figure (a) presents the results of the lattice parameters for individual unit cells of hydrogenated Si46, C6Si40, Ba8Al8Si30, and Ba8Cu8Si38 as a function of volumetric capacity and assuming crystallographic densities. The corresponding comparison for individual unit cells of hydrogenated Si46, C6Si40, Na8Al8Si30, and Li8Al8Si38 are presented in Figure (b). From Figure (a,b), it is apparent that all six silicon-based clathrates considered in this paper (i.e. Si46, C6Si40, Ba8Al8Si30, Ba8Cu8Si38, Na8Al8Si30, and Li8Al8Si38) are promising candidates exhibiting volumetric capacities that may achieve or exceed the DOE targets for vehicles for 2020 and beyond. Furthermore, the Ba-, Na-, and Li-stabilized alloyed clathrates exhibit larger lattice parameters and expand to a greater extent compared to hydrogenated Si46 and C6Si40 at equivalent volumetric capacity. This finding suggests that a framework structure with a large lattice parameter may be beneficial for the volumetric capacity of hydrogen storage.
Figure 5. Predicted volumetric capacity and lattice constant for intermetallic clathrates: (a) hydrogenation of Si46, C6Si40, Ba8Al8Si38, and Ba8Cu8Si38 and (b) hydrogenation of Si46, C6Si40, Na8Al8Si38, and Li8Al8Si38. Vertical lines mark the system-level targets prescribed by DOE.
The DFT computations indicate that for Type I intermetallic clathrate compositions of the formula AxMyX46−y (A = Ba, Na, or Li; M = Al or Cu; and X = Si or Si + C), the internal binding energies for physisorbed hydrogen molecules fall within the energy range desired for reversible storage at or near room temperature (0.1–0.6 eV/H2) as shown in Figure . The DFT computations further show that these Type I intermetallic clathrates can accommodate a large number of hydrogen guest molecules, equivalent to 10 wt.%, and such hydrogenated structures [Ax(H2)zMyX46−y] occur with only a modest increase in lattice volume, as shown in Figures and . These findings are qualitatively in agreement with previous experimental investigations by Neiner et al. [Citation4,Citation5] who showed that hydrogen encapsulation in Na-stabilized and K-stabilized Type I silicon clathrates, resulting in the formation of Na5.5(H2)2.15Si46 and K7(H2)3Si36.
The finding that the volumetric capability increases with increasing lattice parameters may be understood on the basis that the internal void space within the 20-atom and 24-atom cages of the Type I clathrate structure scales with the lattice parameter. Besides having a lager lattice parameter, Al- and Cu-substitution on the framework appears to allow the framework to expand and accommodate more H2 molecules and thus provide enhanced gravimetric and volumetric capacities compared to a pure Si framework or one with C-substitution. The presence of guest atoms such as Ba, Na, and Li do not appear to reduce H2 storage capacity, but allow the binding energy to be tuned to the desirable range of 0.1–0.6 eV/H2.
The current investigation examined only the feasibility of silicon-based clathrates as a hydrogen storage material solely from an energetics consideration without considering the kinetics of hydrogen physisorption and desorption.
The surfaces of the silicon clathrate framework comprised pentagonal and hexagonal rings with different opening sizes for hydrogen entries and releases. Depending on the orientation of the H2 molecules, the entry of an H2 molecule through a five-membered ring or a six-membered ring is different with different activation energy values [Citation9]. Thus, the possibility exists for tailoring the size of the opening of the pentagon and hexagon rings to lower the activation energy barrier through a judicious choice of substitutional elements to replace Si atoms on the framework.
The computational results presented in this work at the upper boundaries of hydrogen insertion are not taken at face value, but rather they are used to establish trends in properties that may point to promising structural and elemental components. From an engineering viewpoint, however, it is clear that the weight and volume savings that can be realized from a storage system designed for physisorption at room temperature and modest pressures (≪ 75 MPa), as opposed to the heavier and more voluminous cryogenic systems presently contemplated for liquid hydrogen or MOF-based solid-state storage, should compensate in part for any potential shortfalls in material storage capacity.
First-principles computational methods
First-principles DFT code VASP [Citation8] was used to calculate the energy of formation, optimal lattice constants of the silicon-based clathrates with guest atoms such as Ba, Na, and Li and substitutional atoms such Al, Cu, and C on the framework. The PBE functional [Citation10] and projector-augmented wave [Citation11] potentials were used along with the plane wave basis sets for the geometry optimization and self-consistent total energy calculations. The energy cut-off for the plane wave basis set was 400 eV. The convergence criteria for energy and forces were set to be 0.01 and 0.1 meV, respectively. Si 3s3p, C 2s2p, Ba 5s5p6s, Li 1s2s, Na 2p3s, Al 3s3p, and Cu 3d4s electrons were treated as valence electrons. Reciprocal space was sampled using 3 × 3 × 3 Monkhorst Pack meshes centred at Gamma. To determine the optimized lattice constants for the alloyed clathrates, the ion positions were relaxed and the volume of the unit cell was optimized. The final lattice constant obtained from the optimized volume was further crosschecked to ensure the pressure in all three x, y, and z are nearly zero.
The formation energies were calculated by subtracting the total energies of the elements from the energy of the structure, then dividing by the total number of atoms. The formation energy, ΔEf, for AxCySi46−y was calculated using the equation given by [Citation6,Citation7]
(1)
where E(Si), E(C), and E(A) are the energies per atom for Si, C (diamond), and A metal, respectively. Using the approach utilized for computing the formation energy of Si with Li to form LixSi [Citation12], the energy of formation of the hydrogenated silicon-based clathrate is computed as
(2)
where z is the number of H2 inserted in the cage and E(H) is the energy of H in molecular H2. Once the formation energy values are computed, the binding energy of H2 in the clathrate system is computed as
(3)
where ΔEb is the binding energy of H2 in the clathrate structure. Equation (3) gives the binding energy per H2 molecule per unit cell since all H2 molecules are encapsulated within the internal cavity of the 3D clathrate structure. The binding energy of the H2 molecules adhered to a 2D sheet of atoms has been defined differently to include H2 molecules that are adhered above and below the 2D layer [Citation13]. External H2 molecules residing outside the 3D clathrate cage structure are not considered in this investigation and Equation (3) is valid as the H2 binding energy. The activation energy for the transport of an H2 molecule into the clathrate cage structure has been investigated by Patterson [Citation9], and is not the subject of this investigation.
The Si-based clathrate compounds that have been selected for studies include empty Si46, empty C6Si40, Al-substituted clathrates with Li, Na, or Ba guest atoms, and Cu-substituted clathrate with Ba guest atoms. The number of hydrogen (H2) molecules inserted into the clathrate framework structure ranged from 0 to 128 in multiples of 8. The hydrogen molecules were placed at randomly selected sites within the cage structure. All the atoms were allowed to relax into equilibrium positions in order to compute the binding energy and the lattice parameter of the unit cell. The formation energy and binding energy were computed without considering the van der Waals interaction between the host and H2. To check this assumption, van der Waals interactions were considered for C6Si40 and Li8Al8Si38 using the DFT-D2 method. Presented in Table S1, the results indicate that van der Waals interaction reduces the binding energy of hydrogenated C6Si40 and Li8Al8Si38 by −0.2657 and −0.2933 eV, respectively. These changes would shift the computed data points toward the lower ΔEb limit but most of the points still remain within the desirable range of 0.1–0.6 eV.
Supplementary_Material
Download MS Word (15.9 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- US Drive Target Explanation Document. Onboard hydrogen storage for light-duty fuel cell vehicles. Washington, DC: Department of Energy; 2015 May.
- Miller MA, Page RA. Standardized testing program for solid-state hydrogen storage technologies. Final report for the Department of Energy hydrogen and fuel cells program, Contract No. DEFC3602AL67619. San Antonio, TX: Southwest Research Institute; 2012 July 30.
- Furukawa H, Miller MA, Yaghi OM. Independent verification of the saturation hydrogen uptake in MOF-177 and establishment of a benchmark for hydrogen adsorption in metal–organic frameworks. J Mater Chem. 2007;17:3197–3204. doi: 10.1039/b703608f
- Neiner D, Okamoto NL, Yu P, et al. Synthesis and characterization of K8−x(H2)ySi46. Inorg Chem. 2010;49(3):815–822. doi: 10.1021/ic9004592
- Neiner D, Okamoto NL, Condron CL, et al. Hydrogen encapsulation in a silicon clathrate type I structure: Na5.5(H2)2.15Si46: synthesis and characterization. J Am Chem Soc. 2007;129(45):13857–13862. doi: 10.1021/ja0724700
- Chan KS, Miller MA, Liang W, et al. First-principles computational design and synthesis of hybrid carbon–silicon clathrates. J Mater Sci. 2014;49(7):2723–2733. doi: 10.1007/s10853-013-7973-6
- Chan KS, Miller MA, Liang W, et al. Computational design and synthesis of nitrogen-substituted carbon and silicon clathrates. Mater Res Lett. 2014;2:70–75. doi: 10.1080/21663831.2013.866988
- Kresse G, Marsman M, Furthmüller J. Vienna ab-initio simulation package − VASP the guide. Wien: Universität Wien; 2012.
- Patterson LSK. Hydrogen storage in silicon clathrates [master’s thesis]. Golden (CO): Colorado School of Mines; 2011.
- Perdew P, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77(18):3865–3868. doi: 10.1103/PhysRevLett.77.3865
- Kresse G, Joubert D. From ultrasoft pseudo potentials to projector augmented-wave method. Phys Rev B. 1999;59(3):1758–1775. doi: 10.1103/PhysRevB.59.1758
- Kubota Y, Escano MCS, Nakanishi H, et al. Austria crystal and electronic structure of Li15Si4. J Appl Phys. 2007;102:053704. doi: 10.1063/1.2775999
- Lei XL, Liu G, Wu MS, et al. Hydrogen storage on calcium-decorated BC7 sheet: a first-principles study. Int J Hydrogen Energy. 2014;39:2142–2148. doi: 10.1016/j.ijhydene.2013.11.099