
Abstract
Nucleotide Excision Repair (NER) is a pathway that removes lesions distorting the DNA helix. The molecular basis of the rare diseases Xeroderma pigmentosum (XP) and Cockayne Syndrome (CS) are explained based on the defects happening in 2 NER branches: Global-Genome Repair and Transcription-Coupled Repair, respectively. Nevertheless, both afflictions sporadically occur together, giving rise to XP/CS; however, the molecular basis of XP/CS is not understood very well. Many efforts have been made to clarify why mutations in only 4 NER genes, namely XPB, XPD, XPF and XPG, are the basis of this disease. Effort has also been made to unravel why mutations within these genes lead to XP, XP/CS, or other pathologies. We have recently contributed to the disclosure of this puzzle by characterizing Rad3/XPD mutations in Saccharomyces cerevisiae and human cells. Based on our, and others', observations, we propose a model compatible with all XP/CS cases and the current bibliography.
Abbreviations
| CS | = | Cockayne Syndrome |
| NER | = | Nucleotide Excision Repair |
| ssDNA | = | single stranded DNA |
| dsDNA | = | double stranded DNA |
| GGR | = | Global Genome Repair |
| TCR | = | Transcription-Coupled Repair |
| XP | = | Xeroderma pigmentosum |
| HR | = | Homologous Recombination |
| rem | = | recombination and mutation |
| CAK | = | Cyclin-Activated Kinase |
| UV | = | Ultraviolet Light |
| DSB | = | Double Strand Break. |
Introduction
Xeroderma pigmentosum (XP) and Cockayne syndrome (CS) are rare autosomal inherited recessive disorders. XP is characterized by cutaneous symptoms because of exposure to sunlight, pigmentation abnormalities, skin atrophy and a high incidence of skin cancer (up to 10000-fold increase)Citation1 and mucous membrane cancer due to the defect in repair of UV- or carcinogen-induced lesions. About 20% of XP patients also display neurological defects that could be explained by neuronal degeneration caused by defects in repair of oxidative damage in the brain region. CS patients display severe growth failure and neurological dysfunction due to demyelization, as well as cachectic dwarfism, ocular abnormalities, sensorineural deafness, dental cavities, microcephaly, skeletal abnormalities, intracranial calcification and mental retardation (For a review see refs.Citation2, 3). In addition, there is a small group of patients that exhibits XP/CS, a combination of clinical symptoms of the 2 diseases.
At the cellular level, the XP and CS conditions are both associated with defects in Nucleotide Excision Repair (NER). This highly orchestrated repair mechanism detects helical distortions as well as DNA thermodynamic destabilizations, signaling them as DNA lesions by specialized proteins. This drives the binding of a protein complex named TFIIH, which allows DNA unwinding, damaged DNA strand recognition and recruitment of specific nucleases that excise the damaged DNA segment. Finally, DNA synthesis fills the remaining single-stranded DNA (ssDNA) gap. The XP molecular basis is explained by defects in Global-Genome Repair (GGR), in which lesions are recognized all over the genome, whereas the molecular basis of CS has been classically explained by defects in Transcription-Coupled Repair (TCR), a NER subpathway in which the lesions are encountered and signaled by the elongating RNA polymerase II, and thus more efficiently repaired in the transcribed DNA strand.Citation4
The XP/CS paradox
A dysfunction exclusively associated with NER during transcription, as that linked to CS without XP manifestation, is due to defects in the proteins CSA and CSB that recognize the lesion at the transcribed site and promote the recruitment of the rest of the NER proteins. However, there are mutations in the XPB, XPD, XPF or XPG genes associated with either XP-only or XP/CS syndromes. On top of that, all mutations described for XPA only confer XP syndrome. Since all the mentioned factors are important for the same repair pathway, it becomes difficult to dissect the molecular basis of XP/CS.
The characterization of a particular subset of mutants of the yeast Saccharomyces cerevisiae has proven useful to start understanding this puzzle.Citation5 We have further worked with mutations of the Rad3/XPD helicase, central to the above-mentioned TFIIH complex. This helicase hydrolyses ATP to open the DNA around the NER lesion so that it can be removed. However, TFIIH also has a fundamental role in the initiation of transcription, since it allows promoter opening via another helicase, Rad25/XPB, and promotes RNA polymerase escape to start elongation.Citation6 The latter function is achieved by the kinase activity of the CDK Activating Kinase (CAK), a subcomplex of TFIIH. In transcription, the Rad3/XPD role is strictly structural, serving to bridge the CAK with the rest of the complex.Citation6,7 Fundamentally, rad3 mutants can be ascribed either to those being highly UV-sensitive, because of a NER defect, and those bearing transcription initiation problems, due to a defect in the TFIIH assembly. We have used a battery of rad3 mutants whose main feature was none of the above: irrespective of their UV sensitivity and without any apparent transcription defect, they require for survival an a priori unrelated DNA repair system, homologous recombination (HR), to be intact. These rad3 mutants are named rem (for increased recombination and mutation levels)Citation8 and they all share the feature of harbouring their mutation in the ATP-binding groove of the protein.Citation9 This fact was highly intriguing since all reported XP/CS patients linked to an XPD deficiency bear their mutations within this same domain.
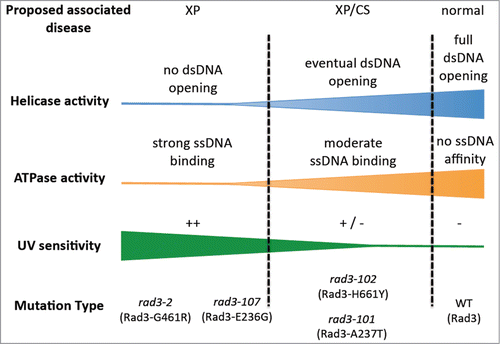
To understand the molecular defect occurring in these mutants we envision the ATPase activity of Rad3 during NER as 2 sides of a coin: it becomes critical for its helicase ability, but it conditions affinity of the protein for ssDNACitation9 (). On this premise, when the mutations in the ATP-binding groove affect it so dramatically that no helicase activity at all can be exerted, the DNA will remain unwound and the TFIIH complex harbouring the mutant Rad3 will float away leaving the lesion unrepaired.Citation10 This should lead to high UV sensitivity, that is, mimicking an XP phenotype. On the contrary, if the mutation has a weak effect that allows DNA melting, once bound, it will promote a stronger binding of the TFIIH complex to the opened DNA.Citation5 This scenario will not only allow damage excision (at least to a certain extent) but it will affect the on-time recruitment of the DNA synthesis machinery,Citation11 leaving a long-lasting ssDNA repair intermediate. In non-cycling cells this may lead to extensive resection of the processed DNA strand, subsequent accumulation of ssDNA and therefore genetic instability.Citation12 In dividing cells, the ssDNA gap would be converted into a double strand break (DSB) when an incoming replication fork hits the unfinished NER intermediate.Citation5 This replication-born DSB requires repair via HR. In addition, the longer persistence of the TFIIH complex at the damaged sites would prevent it from performing new rounds of transcription initiation at promoters, explaining why transcription defects may arise after DNA damage. Altogether, this cascade of events would explain an XP/CS-like phenotype. In this view, the transcription-related defects would be TCR-independent, and therefore serve to explain the paradox presented above. Our model is in agreement with recent findings in various different models: recreation of mutations in the ATP-binding groove of Sulfolobus acidocaldarius XPD leads both to a loss of helicase activity and a gain of affinity for ssDNA;Citation13 in mice, an XP-D/CS-mimicking mutation has been shown to provoke an unfinished NER intermediate that leads to extensive accumulation of ssDNA and subsequent transcription inhibition;Citation14 in XP-D/CS patients cells, the inhibition of transcription after UV is not general but only affects those genes whose promoters were specifically abandoned by TFIIH in response to the damage. Moreover, this correlates with heterochromatinization of the same promoters, tentatively because the retour of the complex is delayed.Citation15 A gradient of phenotypes is expected considering that different mutations in the ATP-binding groove of Rad3/XPD will confer different degrees of helicase inactivation and different levels of gain of affinity for ssDNA, which would explain the various outcomes in the distinct patients.
Figure 1. Proposed molecular defects in Rad3 ATP-binding groove mutants that may help understand XP/CS. The severity of the ATPase defect of rad3/XPD mutants would correlate with both its incapacity of DNA bubble opening and its ssDNA affinity. Mutants more affected were presumed to be the most UV-sensitive, rad3–2 and rad3–107, whereas rad3–101 and rad3–102 would display an intermediate defect, thus allowing eventual opening of the damaged DNA and a major permanence of the TFIIH complex at the DNA.
CAK dissociation from TFIIH as a marker of XP/CS
One of the approaches we have undertaken in order to obtain some idea of the molecular basis of XP/CS has been Fluorescence Recovery After Photo-bleaching (FRAP) in S. cerevisiae Rad3 ATP-binding groove mutants.Citation9 After UV irradiation, the percentage of the fluorescence in the bleached area that cannot be recovered indicates the fraction of TFIIH that is engaged somewhere else performing repair. The time in which the given recovery is achieved is proportional to the molecular weight of the complex being analyzed.Citation16 When the latter quantification was done (by plotting the data relative to a normalized maximum value), we perceived differences in the slopes among the various strains, indicative of faster TFIIH mobility in the mutants down to reaching the WT situation, in which mobility was the slowestCitation9 (). The TFIIH mobility curve, expressed as the mean of the different states of association of a protein with its different partners, had indeed been characterized for a given TFIIH protein.Citation17 In these assays, the steepest slope would mean a higher fraction of assembled TFIIH complexes with lower molecular weight (rad3–2 mutant (Rad3-G461R)). Still lower TFIIH molecular weight within the total TFIIH population would show up in the other mutants (with intermediately steep slopes), while the amount of TFIIH low molecular weight would be minimal in the WT strain (). Interestingly, the module whose dissociation would diminish low molecular weight TFIIH could be the CAK subcomplex. The CAK subcomplex is released from the core TFIIH during NER, which is known to stimulate incision and repair of damaged DNA, while its re-association does not occur until resumption of transcription.Citation18
Figure 2. TFIIH dynamics in Rad3 ATP-binding groove mutants. Representation of FRAP curves normalized to a global maximum of Tfb4-yGFP strains after 80 J/m2 UV-C irradiation. Recovery curves of the normalized fluorescence in wild-type (WT) and rad3 mutants affected in the ATP-binding groove of Rad3 (rad3–102 [Rad3-H661Y], rad3–101 [Rad3-A237T], rad3–107 [Rad3-E236G] and rad3–2 [Rad3-G461R]) are displayed (adapted from data in ref.Citation9). The different steepness of the linear part of the curves shows the average molecular weight of the TFIIH complex under measurement.
![Figure 2. TFIIH dynamics in Rad3 ATP-binding groove mutants. Representation of FRAP curves normalized to a global maximum of Tfb4-yGFP strains after 80 J/m2 UV-C irradiation. Recovery curves of the normalized fluorescence in wild-type (WT) and rad3 mutants affected in the ATP-binding groove of Rad3 (rad3–102 [Rad3-H661Y], rad3–101 [Rad3-A237T], rad3–107 [Rad3-E236G] and rad3–2 [Rad3-G461R]) are displayed (adapted from data in ref.Citation9). The different steepness of the linear part of the curves shows the average molecular weight of the TFIIH complex under measurement.](/cms/asset/b5c1549b-f989-4eb2-8ea5-f126d36519f8/krad_a_1079362_f0002_oc.gif)
A relevant consequence of having an excess of free CAK is an alteration in cell cycle progression, since many key proteins controlling cell cycle stages are targets of the CAK kinase Cdk7.Citation19 Thus, in Drosophila embryos, excess of XPD titrates CAK activity resulting in defects at mitosis, while down regulation of XPD leads to an excess of free CAK and a burst in proliferation.Citation20 In agreement, the yeast mutants rad3–102 (Rad3-H661Y) and rad3–2 show faster entry into the S-phase.Citation5, 21 Altogether, these observations would suggest that an excess of free CAK is a feature of XP-D/CS cells. Indeed, the idea of an excess of free CAK has already been proposed as a molecular hallmark of all XP/CS pathologies.Citation22,23 The proposals were raised because an excess of free CAK had been reported in different XP/CS cells, as is the case of XP-G/CS cells,Citation24 and in XP-D/CS cells, which furthermore show aberrant chromosome segregation due to premature commitment to mitosis.Citation25
An alternative molecular defect for the XP/CS condition
Despite all previous considerations, some observations were noted in the past that do not fit the proposal that excess free CAK is the molecular hallmark of XP/CS.Citation22 First, an XP-B/CS-causing mutation (XPCS1BA patient) was not seen to induce a relevant increase in free CAKCitation24 and, second, mutations in the C-terminus of XPD (XPD-R683W) weakening the association of the CAK to TFIIH were at the base of an XP, but not of an XP/CS, condition.Citation26 One simple way of resolving these inconsistencies would be that the excess of free CAK might just be one possible outcome in certain XP/CS conditions, though not the molecular, causative hallmark at the base of this pathology.
In order to solve these apparent conflicts, and sustain our proposal on the available molecular characterizations,Citation9,13,14 we propose that the molecular feature common to all XP/CS conditions is the persistence of bound TFIIH at an open bubble and the inability to recruit the DNA synthesis factors, which in turn extends the permanence of an unfinished NER intermediate (). This would impair accurate completion of damage repair and sequester TFIIH away from transcription sites impairing transcription resumption, which could explain the XP and CS clinical manifestations.
Figure 3. A possible unified model to explain the XP/CS molecular defect. In a normal situation, TFIIH is recruited to the damaged site, releasing the CAK subcomplex and allowing DNA unwinding and excision of the DNA containing the lesion by the XPF and XPG endonucleases. After DNA repair completion, TFIIH is relocated to the promoter sites, thus allowing transcription resumption. In XP/CS cells, TFIIH is also recruited to the damaged site and performs bubble opening. Incision by XPF or XPG nucleases could be prevented. In all cases, TFIIH could be retained in the DNA as a long-lasting NER intermediate. As a consequence, DNA synthesis would be impaired and DNA break formation may occur whenever incision has happened. TFIIH retention would provoke a delay in transcription resumption. In this scenario, an excess of free CAK is a possible outcome of some XP/CS mutations. Proteins whose deficiencies can cause an XP/CS condition are highlighted in red: XPB, XPD, XPF and XPG.
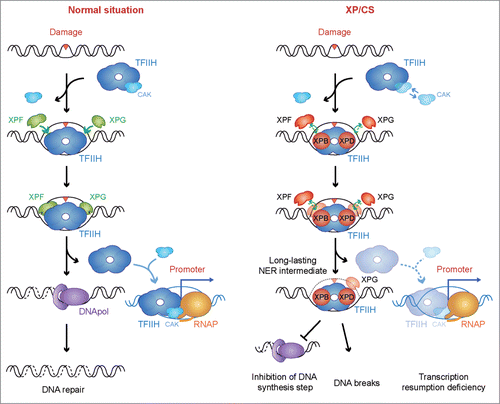
Our model thus provides a uniform explanation compatible with the role during NER not only of XPD (see above), but also of XPB, XPF and XPG. XPB-associated ATPase activity needs to be exerted for NER.Citation27 In analogy with XPD, any loss of ATPase activity in context of a functional TFIIH may cause a gain of affinity for DNA.Citation28 The recently uncovered patient whose XP/CS condition was associated with an XPF deficiencyCitation29 can also be described on the basis of an opened NER bubble without further incision. As for XP-G/CS-causing mutations, it has been shown that they prevent the XPG interaction with TFIIH and may even cause CAK and XPD dissociation.Citation24 TFIIH association with the lesion may therefore occur, but the inability to recruit XPG halts incision by both XPF-ERCC1 and XPG at the 5′ side or at the 3′ side of the lesion, respectively,Citation22,30 and may even cause late XPD and CAK dissociation, leaving an aberrant repair bubble without further processing.Citation24 There is evidence supporting this hypothesis. First, XPB mutations leading to XP/CS have been shown to fully allow TFIIH recruitment and subsequent assembly of the incision nucleases,Citation11 which is accompanied by bubble opening and XPG incision. Since this XPB mutation impairs XPF incision,Citation31 the NER intermediate is long lastingCitation32 and DNA synthesis does not take place.Citation11 Second, XPG mutations leading to XP/CS are as well able to promote TFIIH assembly and bubble establishment, but recruitment of DNA synthesis factors is prevented.Citation11 Altogether, we propose the long-lasting NER intermediate as the general molecular defect underlying all XP/CS deficiencies, irrespective of whether it is XPD, XPB, XPF or XPG being mutated (). As a consequence, and only in some circumstances, this may lead to an excess of free CAK.
Therapeutic possibilities of the model
Some therapeutic considerations could be derived from our proposal. If the mutations at the base of XP/CS are causing the molecular defect we have described here, namely a long-lasting NER intermediate, this could be exploited for XP/CS-associated cancer treatment. As previously explained, the impact of these mutations would be different in non-dividing versus cycling cells. The latter would present an acute dependency on HR functions in order to tolerate the unfinished NER. In the absence of HR, DSBs would accumulate in their genomes, eventually leading to cell death. Given that cancer cells cycle more frequently than most body cells, inhibition of HR could be explored as a therapeutic approach to XP/CS-associated cancers. Moreover, not only the spontaneous damage that is a substrate of NER would generate DSBs in an XP/CS-like context, but also their production could be exacerbated if cells are directly challenged with UV light-mimetic drugs.Citation9 Thus, if the local delivery of such drugs in combination with HR inhibitors is feasible, this could be investigated as a way to hyper-sensitize XP/CS cancer cells.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank D Haun for style supervision.
Funding
Research in AA's lab is funded by grants from the Spanish Ministry of Economy and Competitiveness, the Junta de Andalucía and the European Union (FEDER).
References
- Bradford PT, Goldstein AM, Tamura D, Khan SG, Ueda T, Boyle J, Oh KS, Imoto K, Inui H, Moriwaki S, et al. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet 2011; 48:168-76; PMID:21097776; http://dx.doi.org/10.1136/jmg.2010.083022
- Cleaver JE, Lam ET, Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet 2009; 10:756-68; PMID:19809470; http://dx.doi.org/10.1038/nrg2663
- de Boer J, Hoeijmakers JH. Nucleotide excision repair and human syndromes. Carcinogenesis 2000; 21:453-60; PMID:10688865; http://dx.doi.org/10.1093/carcin/21.3.453
- Venema J, Mullenders LH, Natarajan AT, van Zeeland AA, Mayne LV. The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc Natl Acad Sci U S A 1990; 87:4707-11; PMID:2352945; http://dx.doi.org/10.1073/pnas.87.12.4707
- Moriel-Carretero M, Aguilera A. A postincision-deficient TFIIH causes replication fork breakage and uncovers alternative Rad51- or Pol32-mediated restart mechanisms. Mol Cell 2010; 37:690-701; PMID:20227372; http://dx.doi.org/10.1016/j.molcel.2010.02.008
- Tirode F, Busso D, Coin F, Egly JM. Reconstitution of the transcription factor TFIIH: assignment of functions for the three enzymatic subunits, XPB, XPD, and cdk7. Mol Cell 1999; 3:87-95; PMID:10024882; http://dx.doi.org/10.1016/S1097-2765(00)80177-X
- Kuper J, Braun C, Elias A, Michels G, Sauer F, Schmitt DR, Poterszman A, Egly JM, Kisker C. In TFIIH, XPD helicase is exclusively devoted to DNA repair. PLoS Biol 2014; 12:e1001954; PMID:25268380; http://dx.doi.org/10.1371/journal.pbio.1001954
- Montelone BA, Hoekstra MF, Malone RE. Spontaneous mitotic recombination in yeast: the hyper-recombinational rem1 mutations are alleles of the RAD3 gene. Genetics 1988; 119:289-301; PMID:2840336
- Herrera-Moyano E, Moriel-Carretero M, Montelone BA, Aguilera A. The rem mutations in the ATP-binding groove of the Rad3/XPD helicase lead to Xeroderma pigmentosum-Cockayne Syndrome-Like phenotypes. PLoS Genet 2014; 10:e1004859; PMID:25500814; http://dx.doi.org/10.1371/journal.pgen.1004859
- Naumovski L, Chu G, Berg P, Friedberg EC. RAD3 gene of Saccharomyces cerevisiae: nucleotide sequence of wild-type and mutant alleles, transcript mapping, and aspects of gene regulation. Mol Cell Biol 1985; 5:17-26; PMID:3885009
- Mocquet V, Laine JP, Riedl T, Yajin Z, Lee MY, Egly JM. Sequential recruitment of the repair factors during NER: the role of XPG in initiating the resynthesis step. EMBO J 2008; 27:155-67; PMID:18079701; http://dx.doi.org/10.1038/sj.emboj.7601948
- Giannattasio M, Follonier C, Tourriere H, Puddu F, Lazzaro F, Pasero P, Lopes M, Plevani P, Muzi-Falconi M. Exo1 competes with repair synthesis, converts NER intermediates to long ssDNA gaps, and promotes checkpoint activation. Mol Cell 2010; 40:50-62; PMID:20932474; http://dx.doi.org/10.1016/j.molcel.2010.09.004
- Fan L, Fuss JO, Cheng QJ, Arvai AS, Hammel M, Roberts VA, Cooper PK, Tainer JA. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell 2008; 133:789-800; PMID:18510924; http://dx.doi.org/10.1016/j.cell.2008.04.030
- Godon C, Mourgues S, Nonnekens J, Mourcet A, Coin F, Vermeulen W, Mari PO, Giglia-Mari G. Generation of DNA single-strand displacement by compromised nucleotide excision repair. EMBO J 2012; 31:3550-63; PMID:22863773; http://dx.doi.org/10.1038/emboj.2012.193
- Velez-Cruz R, Zadorin AS, Coin F, Egly JM. Sirt1 suppresses RNA synthesis after UV irradiation in combined xeroderma pigmentosum group D/Cockayne syndrome (XP-D/CS) cells. Proc Natl Acad Sci U S A 2013; 110:E212-20; PMID:23267107; http://dx.doi.org/10.1073/pnas.1213076110
- Goodwin JS, Kenworthy AK. Photobleaching approaches to investigate diffusional mobility and trafficking of Ras in living cells. Methods 2005; 37:154-64; PMID:16288889; http://dx.doi.org/10.1016/j.ymeth.2005.05.013
- Giglia-Mari G, Miquel C, Theil AF, Mari PO, Hoogstraten D, Ng JM, Dinant C, Hoeijmakers JH, Vermeulen W. Dynamic interaction of TTDA with TFIIH is stabilized by nucleotide excision repair in living cells. PLoS Biol 2006; 4:e156; PMID:16669699; http://dx.doi.org/10.1371/journal.pbio.0040156
- Coin F, Oksenych V, Mocquet V, Groh S, Blattner C, Egly JM. Nucleotide excision repair driven by the dissociation of CAK from TFIIH. Mol Cell 2008; 31:9-20; PMID:18614043; http://dx.doi.org/10.1016/j.molcel.2008.04.024
- Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci 2005; 118:5171-80; PMID:16280550; http://dx.doi.org/10.1242/jcs.02718
- Chen J, Larochelle S, Li X, Suter B. Xpd/Ercc2 regulates CAK activity and mitotic progression. Nature 2003; 424:228-32; PMID:12853965; http://dx.doi.org/10.1038/nature01746
- Moriel-Carretero M, Aguilera A. Replication fork breakage and re-start: New insights into Rad3/XPD-associated deficiencies. Cell Cycle 2010; 9:2958-62; PMID:20740714; http://dx.doi.org/10.4161/cc.9.15.12408
- Scharer OD. Hot topics in DNA repair: the molecular basis for different disease states caused by mutations in TFIIH and XPG. DNA Repair (Amst) 2008; 7:339-44; PMID:18077223; http://dx.doi.org/10.1016/j.dnarep.2007.10.007
- Arab HH, Wani G, Ray A, Shah ZI, Zhu Q, Wani AA. Dissociation of CAK from core TFIIH reveals a functional link between XP-G/CS and the TFIIH disassembly state. PLoS One 2010; 5:e11007; PMID:20543986; http://dx.doi.org/10.1371/journal.pone.0011007
- Ito S, Kuraoka I, Chymkowitch P, Compe E, Takedachi A, Ishigami C, Coin F, Egly JM, Tanaka K. XPG stabilizes TFIIH, allowing transactivation of nuclear receptors: implications for Cockayne syndrome in XP-G/CS patients. Mol Cell 2007; 26:231-43; PMID:17466625; http://dx.doi.org/10.1016/j.molcel.2007.03.013
- Ito S, Tan LJ, Andoh D, Narita T, Seki M, Hirano Y, Narita K, Kuraoka I, Hiraoka Y, Tanaka K. MMXD, a TFIIH-independent XPD-MMS19 protein complex involved in chromosome segregation. Mol Cell 2010; 39:632-40; PMID:20797633; http://dx.doi.org/10.1016/j.molcel.2010.07.029
- Keriel A, Stary A, Sarasin A, Rochette-Egly C, Egly JM. XPD mutations prevent TFIIH-dependent transactivation by nuclear receptors and phosphorylation of RARalpha. Cell 2002; 109:125-35; PMID:11955452; http://dx.doi.org/10.1016/S0092-8674(02)00692-X
- Fan L, Arvai AS, Cooper PK, Iwai S, Hanaoka F, Tainer JA. Conserved XPB core structure and motifs for DNA unwinding: implications for pathway selection of transcription or excision repair. Mol Cell 2006; 22:27-37; PMID:16600867; http://dx.doi.org/10.1016/j.molcel.2006.02.017
- Winkler GS, Sugasawa K, Eker AP, de Laat WL, Hoeijmakers JH. Novel functional interactions between nucleotide excision DNA repair proteins influencing the enzymatic activities of TFIIH, XPG, and ERCC1-XPF. Biochemistry 2001; 40:160-5; PMID:11141066; http://dx.doi.org/10.1021/bi002021b
- Kashiyama K, Nakazawa Y, Pilz DT, Guo C, Shimada M, Sasaki K, Fawcett H, Wing JF, Lewin SO, Carr L, et al. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. American journal of human genetics 2013; 92:807-19; PMID:23623389; http://dx.doi.org/10.1016/j.ajhg.2013.04.007
- Staresincic L, Fagbemi AF, Enzlin JH, Gourdin AM, Wijgers N, Dunand-Sauthier I, Giglia-Mari G, Clarkson SG, Vermeulen W, Scharer OD. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J 2009; 28:1111-20; PMID:19279666; http://dx.doi.org/10.1038/emboj.2009.49
- Coin F, Auriol J, Tapias A, Clivio P, Vermeulen W, Egly JM. Phosphorylation of XPB helicase regulates TFIIH nucleotide excision repair activity. EMBO J 2004; 23:4835-46; PMID:15549133; http://dx.doi.org/10.1038/sj.emboj.7600480
- Oh KS, Imoto K, Boyle J, Khan SG, Kraemer KH. Influence of XPB helicase on recruitment and redistribution of nucleotide excision repair proteins at sites of UV-induced DNA damage. DNA Repair (Amst) 2007; 6:1359-70; PMID:17509950; http://dx.doi.org/10.1016/j.dnarep.2007.03.025