
ABSTRACT
Duchenne muscular dystrophy (DMD) is the most common and severe inherited neuromuscular disorder. DMD is caused by mutations in the gene encoding the dystrophin protein in muscle fibers. Dystrophin was originally proposed to be a structural protein that protected the sarcolemma from stresses produced during contractions. However, more recently, experimental evidence has revealed a far more complicated picture, with the loss of dystrophin causing dysfunction of multiple muscle signaling pathways, which all contribute to the overall disease pathophysiology. Current gene-based approaches for DMD are conceptually appealing since they offer the potential to restore dystrophin to muscles, albeit a partially functional, truncated form of the protein. However, given the cost and technical challenges facing these genetic approaches, it is important to consider if relatively inexpensive, clinically used drugs may be repurposed for treating DMD. Here, we discuss our recent findings showing the potential of simvastatin as a novel therapy for DMD.
Background and current therapeutic approaches in DMD
Duchenne muscular dystrophy (DMD) is classified as a rare (orphan) disease, affecting 1:5000 males worldwide. However, among inherited neuromuscular diseases, DMD is the most common and severe disorder. DMD is caused by mutations in the gene encoding the dystrophin protein, which in most cases results in no detectable dystrophin expression in muscle. Dystrophin is a large (427 kDa) sarcolemmal protein that links the cytoskeleton to a membrane-bound protein complex, the dystrophin-associated protein complex (DPC). For a number of years, dystrophin was thought to provide a mechanical-stabilizing role in muscle fibers, by protecting the sarcolemma from damage during contractions.Citation1 However, particularly over the last decade or so, experimental evidence has revealed far more complex and diverse cellular roles for dystrophin. The loss of dystrophin in DMD and mdx mice (a mouse model of DMD) leads to perturbations in numerous signaling and homeostatic pathways, which contribute in various ways to the ongoing muscle damage and impairment in muscle function.
Despite recent advancements in our understanding of the mechanisms causing DMD, there is still currently no effective long-term treatment for DMD. Therefore, there is an urgent need for finding effective, clinically approved, therapies in the immediate future. The mainstay treatment for more than a decade has been corticosteroids such as prednisone and deflazacort. These drugs do provide some improvement in muscle strength and mobility and delay, by a few years, the time for patients to require a wheelchair.Citation2 Nevertheless, corticosteroids cause significant side-effects, including weight gain and increased bone fracturesCitation3 and therefore are not an ideal long-term treatment option. More recently, gene-based approaches designed to generate a truncated dystrophin protein in DMD muscles have been developed and some ‘exon skipping’ oligonucleotides are currently being tested in clinical trials.Citation4 While these strategies are conceptually appealing, there will be several technical and regulatory hurdles to overcome before they are readily available for DMD patients.Citation5,6 Moreover, in the case of exon skipping, each oligonucleotide is designed to target a specific dystrophin mutation and as such, independent testing on a specific group of patients harboring that mutation will be required.Citation5 This will further delay the time taken for the majority of patients to have access to a potential treatment.
Rationale for evaluating simvastatin as a therapeutic approach for DMD
We have taken a different approach toward developing a potential treatment for DMD. We have focused on repurposing existing pharmacological agents that target specific signaling pathways known to contribute to dystrophic disease progression and the loss of muscle function.Citation7,8 In our recent study, we treated mdx mice with simvastatin, a common statin medication used by millions of people worldwide to treat high circulating LDL cholesterol levels.Citation9 While lowering LDL cholesterol is the primary goal of statin treatment, our rationale for using simvastatin in muscular dystrophy was based on cholesterol-independent, ‘pleiotropic’ benefits of statins. These benefits include reducing oxidative stress, inflammation and fibrosis,Citation10,11 three key pathogenic processes in DMD that are major mediators of functional impairment. In terms of oxidative stress, we and others have recently shown that NADPH oxidase 2 (NOX2) is a major source of reactive oxygen species (ROS) production in mdx muscle.Citation12-15 NOX2 levels are increased in mdx muscle and during stretched contractions, ROS produced by NOX2 triggers opening of a stretch-activated channel (SAC), which causes excessive Ca2+ entry into dystrophic muscle fibers.Citation15 High levels of ROS and Ca2+ produced by this pathway stimulate increased membrane permeability, increase muscle damage and reduce force production.Citation8,16 Since statins are known to reduce NOX2 expression and ROS production in the cardiovascular system,Citation17,18 we were particularly interested to test if simvastatin could inhibit NOX2 in mdx muscle. Finally, we chose simvastatin rather than other statins since it is the most lipophilic, which increases its uptake by peripheral tissues, including skeletal muscle.
The dystrophin complex is not essential for substantial improvements in muscle health and function provided by simvastatin
The results of our study demonstrated that simvastatin provided substantial improvements in overall muscle health and function when administered both short and long-term, and at different stages of the disease.Citation9 In support of our hypothesis, simvastatin provided significant protection against muscle damage, as shown by dramatic reductions in plasma creatine kinase, inflammation, fibrosis and oxidative stress. NOX2 levels were reduced to wild type (WT) control levels in simvastatin-treated mdx mice and this correlated with enhanced muscle force production by the tibialis anterior (TA) muscle. Together, we propose that the inhibition of these pathogenic pathways by simvastatin minimizes muscle damage and enhances muscle function ().
Figure 1. Simvastatin inhibits pathogenic pathways that impair muscle function in dystrophic muscle. Loss of dystrophin increases inflammation, fibrosis, and oxidative stress by NOX2. This leads to muscle damage and progressive weakness. Simvastatin reduces each of these pathways (red arrows), improving muscle health and function.
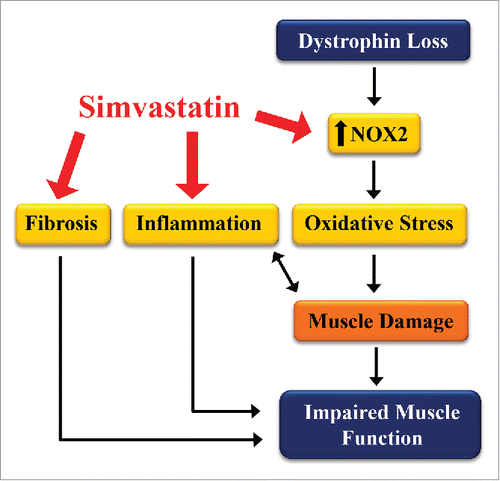
Of particular interest, we demonstrated that restoring dystrophin and/or the DPC is not essential to provide a substantial improvement in dystrophic muscle health and force production. Simvastatin did not increase the expression of the dystrophin homolog, utrophin, or other DPC proteins including β-dystroglycan, α-syntrophin and α-dystrobrevin. Moreover, the increase in TA specific muscle force provided by simvastatin (40%) is comparable to that provided by the most effective mini-dystrophin gene therapy constructCitation19 and ‘exon skipping’ antisense oligonucleotides, which lead to the production of a slightly shorter dystrophin isoform throughout the mdx TA muscle.Citation20 Our data highlight the point that drugs such as simvastatin, designed to target specific pathogenic pathways in dystrophic muscles, can provide functional improvements that compare favorably with the most effective gene-based approaches for DMD.
Simvastatin reverses pre-existing fibrosis in severely dystrophic muscle
Fibrosis is a major cause of progressive muscle weakness in DMD.Citation21 The replacement of muscle with connective tissue impairs muscle strength and ultimately limits mobility, leading to loss of ambulation by about the age of 10.Citation21 Importantly, in terms of choosing the most efficacious therapies for DMD, recent evidence has shown that fibrosis begins very early in the disease, by 1 to 2 y of age, often several years before first diagnosis of the disease.Citation22 At this young age, already 15% of the muscle is comprised of connective tissue as opposed to 3% in healthy boys.Citation22 For DMD, this value increases up to 30% by ages 7 to 10.Citation22 Therefore, in the case of genetic approaches, implementation of early therapeutic intervention will be critical to maximize any potential benefits, since these therapies rely on the presence of viable muscle tissue and are unlikely to reverse advanced fibrosis in dystrophic muscles.
In 12 month old mdx mice, the severely dystrophic diaphragm muscle has substantial fibrosis and force loss, which closely recapitulates the pathology and function of DMD muscles. We showed that simvastatin reversed pre-existing diaphragm fibrosis by 50% in these old mdx mice, as quantified by fibronectin and collagen I levels, and this was accompanied by a significant 20–30% increase in diaphragm force production. The reversal of fibrosis with simvastatin is consistent with results from a hypertrophic cardiomyopathy animal model, which also showed regression of fibrosis following simvastatin treatment.Citation23 Overall, our data suggests that simvastatin could potentially provide benefits to both young and older DMD patients. An important follow up question to these findings is whether the reversal of fibrosis by simvastatin can also create an environment conducive to muscle regeneration that could potentially repopulate the muscle with new myofibers.
Clinical prospects for simvastatin in DMD
To our knowledge, statins have never been tested as a therapy for any inherited muscle disease. This is primarily due to the perceived risk of muscle related symptoms that can occur with statin use. A recent review of several randomized, placebo controlled studies showed that the incidence of adverse, statin-related muscle symptoms was almost identical for statin treated and placebo control patients.Citation24 This data indicates that the overall incidence of statin-induced muscle complaints is actually quite rare and that a significant number of patients taking statins have muscle pain or weakness that is unrelated to statin use. Importantly, long-term statin treatment for hypercholesterolemia in pediatric patients, the age group most relevant for DMD, has been shown to be effective, in terms of LDL cholesterol lowering, and safe, with no evidence of muscle-related side effects.Citation25 Therefore, clinical findings to date suggest that statins are safe for use in the pediatric population.
While statin myopathy is rare, animal studies have identified some specific mechanisms that are involved, including increased mitochondrial ROS productionCitation26 and activation of the atrogin-1 muscle wasting pathway.Citation27 Importantly, we showed that simvastatin decreased oxidative stress in mdx muscle and had no effect on atrogin-1 levels, which were lower in mdx compared to normal WT muscle. This is consistent with findings in DMD muscles, where atrogin-1 levels are consistently lower than those of control at various stages of the disease.Citation28 These data suggest that the underlying disease mechanisms in dystrophic muscle may be both amenable to simvastatin treatment and protective against possible side effects. To emphasize this point, corticosteroids are the most common cause of drug-induced myopathy and atrophy,Citation29 yet paradoxically, in DMD patients these drugs improve muscle function, despite causing the other well-known, metabolism-related steroid side-effects. While it is true that findings from preclinical, animal studies often do not translate directly to humans, a large number of clinical studies using simvastatin and other statins have shown that these drugs do provide significant clinical benefits in a wide-range of human diseases. Using fibrosis as an example, a recent large, clinical trial showed a significant reduction in the progression of liver fibrosis in statin-treated patients with chronic hepatitis C.Citation30
Conclusion
In summary, we have shown in a preclinical animal model of DMD that simvastatin, one of the most commonly used medications in the world, provides considerable benefits to the overall health and function of dystrophic muscles. These results highlight the utility of targeting specific pathogenic pathways, oxidative stress, inflammation and fibrosis, which are all major causes of the disease pathophysiology. Given the immediate need for therapies that are amenable to all DMD patients, regardless of their age or specific dystrophin mutation, simvastatin has great potential to provide a cost effective, readily available therapy for DMD.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A 1993; 90:3710-4; PMID:8475120; http://dx.doi.org/10.1073/pnas.90.8.3710
- Bello L, Gordish-Dressman H, Morgenroth LP, Henricson EK, Duong T, Hoffman EP, Cnaan A, McDonald CM, Investigators C. Prednisone/prednisolone and deflazacort regimens in the CINRG Duchenne Natural History Study. Neurol 2015; 85:1048-55; PMID:26311750; http://dx.doi.org/10.1212/WNL.0000000000001950
- Bianchi ML, Biggar D, Bushby K, Rogol AD, Rutter MM, Tseng B. Endocrine aspects of Duchenne muscular dystrophy. Neuromuscul Disord 2011; 21:298-303; PMID:21353552; http://dx.doi.org/10.1016/j.nmd.2011.02.006
- Kole R, Krieg AM. Exon skipping therapy for Duchenne muscular dystrophy. Adv Drug Deliv Rev 2015; 87:104-7; PMID:25980936; http://dx.doi.org/10.1016/j.addr.2015.05.008
- Aartsma-Rus A, Ferlini A, Goemans N, Pasmooij AM, Wells DJ, Bushby K, Vroom E, Balabanov P. Translational and regulatory challenges for exon skipping therapies. Hum Gene Ther 2014; 25:885-92; PMID:25184444; http://dx.doi.org/10.1089/hum.2014.086
- Hoffman EP, McNally EM. Exon-skipping therapy: a roadblock, detour, or bump in the road? Sci Transl Med 2014; 6:230fs14; PMID:24695683; http://dx.doi.org/10.1126/scitranslmed.3008873
- Percival JM, Whitehead NP, Adams ME, Adamo CM, Beavo JA, Froehner SC. Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J Pathol 2012; 228:77-87; PMID:22653783
- Whitehead NP, Pham C, Gervasio OL, Allen DG. N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J Physiol 2008; 586:2003-14; PMID:18258657; http://dx.doi.org/10.1113/jphysiol.2007.148338
- Whitehead NP, Kim MJ, Bible KL, Adams ME, Froehner SC. A new therapeutic effect of simvastatin revealed by functional improvement in muscular dystrophy. Proc Natl Acad Sci U S A 2015; 112:12864-9; PMID:26417069; http://dx.doi.org/10.1073/pnas.1509536112
- Antonopoulos AS, Margaritis M, Shirodaria C, Antoniades C. Translating the effects of statins: from redox regulation to suppression of vascular wall inflammation. Thromb Haemost 2012; 108:840-8; PMID:22872079; http://dx.doi.org/10.1160/TH12-05-0337
- Tanaka S, Fukumoto Y, Nochioka K, Minami T, Kudo S, Shiba N, Takai Y, Williams CL, Liao JK, Shimokawa H. Statins exert the pleiotropic effects through small GTP-binding protein dissociation stimulator upregulation with a resultant Rac1 degradation. Arterioscler Thromb Vasc Biol 2013; 33:1591-600; PMID:23640485; http://dx.doi.org/10.1161/ATVBAHA.112.300922
- Khairallah RJ, Shi G, Sbrana F, Prosser BL, Borroto C, Mazaitis MJ, Hoffman EP, Mahurkar A, Sachs F, Sun Y, et al. Microtubules underlie dysfunction in duchenne muscular dystrophy. Sci Signal 2012; 5:ra56; PMID:22871609; http://dx.doi.org/10.1126/scisignal.2002829
- Pal R, Palmieri M, Loehr JA, Li S, Abo-Zahrah R, Monroe TO, Thakur PB, Sardiello M, Rodney GG. Src-dependent impairment of autophagy by oxidative stress in a mouse model of Duchenne muscular dystrophy. Nat Commun 2014; 5:4425; PMID:25028121
- Shkryl VM, Martins AS, Ullrich ND, Nowycky MC, Niggli E, Shirokova N. Reciprocal amplification of ROS and Ca(2+) signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Arch 2009; 458:915-28; PMID:19387681; http://dx.doi.org/10.1007/s00424-009-0670-2
- Whitehead NP, Yeung EW, Froehner SC, Allen DG. Skeletal muscle NADPH oxidase is increased and triggers stretch-induced damage in the mdx mouse. PLoS One 2010; 5:e15354; PMID:21187957; http://dx.doi.org/10.1371/journal.pone.0015354
- Whitehead NP, Streamer M, Lusambili LI, Sachs F, Allen DG. Streptomycin reduces stretch-induced membrane permeability in muscles from mdx mice. Neuromuscul Disord 2006; 16:845-54; PMID:17005404; http://dx.doi.org/10.1016/j.nmd.2006.07.024
- Dalaklioglu S, Sahin P, Tasatargil A, Celik-Ozenci C. Pravastatin improves the impaired nitric oxide-mediated neurogenic and endothelium-dependent relaxation of corpus cavernosum in aged rats. Aging Male 2014; 17(4):259-66; PMID:24000938
- Pignatelli P, Carnevale R, Pastori D, Cangemi R, Napoleone L, Bartimoccia S, Nocella C, Basili S, Violi F. Immediate antioxidant and antiplatelet effect of atorvastatin via inhibition of Nox2. Circulation 2012; 126:92-103; PMID:22615342; http://dx.doi.org/10.1161/CIRCULATIONAHA.112.095554
- Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C, Judge L, Bostick B, Chamberlain JS, Terjung RL, et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest 2009; 119:624-35; PMID:19229108; http://dx.doi.org/10.1172/JCI36612
- Godfrey C, Muses S, McClorey G, Wells KE, Coursindel T, Terry R, Betts C, Hammond S, O'Donovan L, Hildyard J, et al. How much dystrophin is enough: the physiological consequences of different levels of dystrophin in the mdx mouse. Hum Mol Genet 2015; 24(15):4225-37
- Desguerre I, Mayer M, Leturcq F, Barbet JP, Gherardi RK, Christov C. Endomysial fibrosis in Duchenne muscular dystrophy: a marker of poor outcome associated with macrophage alternative activation. J Neuropathol Exp Neurol 2009; 68:762-73; PMID:19535995; http://dx.doi.org/10.1097/NEN.0b013e3181aa31c2
- Peverelli L, Testolin S, Villa L, D'Amico A, Petrini S, Favero C, Magri F, Morandi L, Mora M, Mongini T, et al. Histologic muscular history in steroid-treated and untreated patients with Duchenne dystrophy. Neurol 2015; 85(21):1886-93; PMID:26497992
- Patel R, Nagueh SF, Tsybouleva N, Abdellatif M, Lutucuta S, Kopelen HA, Quinones MA, Zoghbi WA, Entman ML, Roberts R, et al. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation 2001; 104:317-24; PMID:11457751; http://dx.doi.org/10.1161/hc2801.094031
- Ganga HV, Slim HB, Thompson PD. A systematic review of statin-induced muscle problems in clinical trials. Am Heart J 2014; 168:6-15; PMID:24952854; http://dx.doi.org/10.1016/j.ahj.2014.03.019
- Braamskamp MJ, Kusters DM, Avis HJ, Smets EM, Wijburg FA, Kastelein JJ, Wiegman A, Hutten BA. Long-term statin treatment in children with familial hypercholesterolemia: more insight into tolerability and adherence. Paediatr Drugs 2015; 17:159-66; PMID:25644328; http://dx.doi.org/10.1007/s40272-014-0116-y
- Bouitbir J, Charles AL, Echaniz-Laguna A, Kindo M, Daussin F, Auwerx J, Piquard F, Geny B, Zoll J. Opposite effects of statins on mitochondria of cardiac and skeletal muscles: a ‘mitohormesis’ mechanism involving reactive oxygen species and PGC-1. Eur Heart J 2012; 33:1397-407; PMID:21775390; http://dx.doi.org/10.1093/eurheartj/ehr224
- Hanai J, Cao P, Tanksale P, Imamura S, Koshimizu E, Zhao J, Kishi S, Yamashita M, Phillips PS, Sukhatme VP, et al. The muscle-specific ubiquitin ligase atrogin-1/MAFbx mediates statin-induced muscle toxicity. J Clin Invest 2007; 117:3940-51; PMID:17992259
- Chen YW, Nagaraju K, Bakay M, McIntyre O, Rawat R, Shi R, Hoffman EP. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurol 2005; 65:826-34; PMID:16093456; http://dx.doi.org/10.1212/01.wnl.0000173836.09176.c4
- Gupta A, Gupta Y. Glucocorticoid-induced myopathy: Pathophysiology, diagnosis, and treatment. Indian J Endocrinol Metab 2013; 17:913-6; PMID:24083177; http://dx.doi.org/10.4103/2230-8210.117215
- Butt AA, Yan P, Bonilla H, Abou-Samra AB, Shaikh OS, Simon TG, Chung RT, Rogal SS, Team ES. Effect of addition of statins to antiviral therapy in hepatitis C virus-infected persons: Results from ERCHIVES. Hepatol 2015; 62:365-74; PMID:25847403; http://dx.doi.org/10.1002/hep.27835