
ABSTRACT
We previously demonstrated elevated brain iron levels in myelinated structures and associated cells in a hemochromatosis Hfe−/−xTfr2mut mouse model. This was accompanied by altered expression of a group of myelin-related genes, including a suite of genes causatively linked to the rare disease family ‘neurodegeneration with brain iron accumulation’ (NBIA). Expanded data mining and ontological analyses have now identified additional myelin-related transcriptome changes in response to brain iron loading. Concordance between the mouse transcriptome changes and human myelin-related gene expression networks in normal and NBIA basal ganglia testifies to potential clinical relevance. These analyses implicate, among others, genes linked to various rare central hypomyelinating leukodystrophies and peripheral neuropathies including Pelizaeus-Merzbacher-like disease and Charcot-Marie-Tooth disease as well as genes linked to other rare neurological diseases such as Niemann-Pick disease. The findings may help understand interrelationships of iron and myelin in more common conditions such as hemochromatosis, multiple sclerosis and various psychiatric disorders.
Introduction
Neurodegeneration with Brain Iron Accumulation (NBIA) comprises a family of rare, often fatal diseases that can present with progressive motor dysfunction such as dystonia, dysarthria and spasticity, visual impairment and behavioral or cognitive problems.Citation1 While iron accumulation in the basal ganglia is a common feature, most of the genes causatively associated with the 10 published NBIA subtypes do not have clear links to iron homeostasis and not all symptomatic patients with NBIA mutations have obvious iron abnormalities, creating uncertainty as to the nature of the relationships between iron accumulation and the clinical and neuropathological features of NBIA.
In the past there have been few appropriate research models for studying the effects of elevated iron in the brain. Animal models have usually entailed short-term iron loading by injection or dietary iron supplementation, with limited relevance to humans, where pathology due to iron accumulation can take several years to manifest clinically, even in the more aggressive iron diseases.Citation2 Although recent years have seen the emergence of genetically modified mouse models for studying brain iron homeostasis, most have not exhibited substantiated increases in brain iron loading.
A new Hfe−/−xTfr2mut mouse model of the common iron loading disease hemochromatosis has now been introduced that has simultaneous disruption of the hemochromatosis Hfe and transferrin receptor 2 Tfr2 genes, on an AKR background, and hepatopathological signs of clinical hemochromatosis.Citation3 We have recently reported significantly increased brain iron loading in these mice relative to wildtype mice, the first such demonstration in a hemochromatosis model, accompanied by subtle behavioral changes, specifically hyperactivity and possibly also increased aggression.Citation4 Hemochromatosis in individuals with combined mutations in both the TFR2 and HFE genes is typically of earlier onset but otherwise clinically indistinguishable from hemochromatosis caused by either of these mutations alone.Citation4
Compared to wildtype mice, Hfe−/−xTfr2mut mice showed higher iron accumulation in myelinated structures throughout the brain, and in oligodendroglial cells and other as yet unidentified cells, with few if any neurons, astrocytes or microglia showing detectable iron staining. Transcriptomic studies suggested that subtle effects on molecular systems related to myelin might be among the earliest pathological sequelae of increased brain iron loading, rather than the neurodegenerative changes iron is usually assumed to cause, such as direct oxidative damage to neurons. This raises the possibility that myelin pathology may provide a “missing link” between abnormal brain iron accumulation and early functional and behavioral sequelae.Citation4
There were altered levels of the iron storage protein, ferritin, linked to the NBIA subtype neuroferritinopathy, and of transcripts for 5 other NBIA-associated genes (ceruloplasmin Cp (MIM117700); phospholipase A2, group VI Pla2g6 (MIM603604); fatty acid 2-hydroxylase Fa2h (MIM611026); chromosome 19 open reading frame 12 C19orf12 (MIM614297) and ATPase type 13A2 Atp13a2 (MIM610513)). Myelin pathology has been reported in NBIA patients or mice with mutations in each of these genes, including the gene encoding ferritin, and we also identified 16 other myelin-related genes with decreased expression in Hfe−/−xTfr2mut brain.Citation4
As discussed in our earlier paper,Citation4 no pathological changes were observed in the gross structure or ultrastructure of myelin. This is as expected, since the Hfe−/−xTfr2mut mice can be considered equivalent to the initial phases of human hemochromatosis, which do not entail clinically significant brain problems, whereas in NBIA severe CNS phenotypes can be present even very early in childhood. Nonetheless it appears likely, in view of the substantial changes in myelin gene expression observed in these mice, that myelin-related molecular systems are affected to some degree. This may potentially increase the likelihood of myelin pathology developing in conjunction with other causes, for example inappropriate immune responses.
As far as we know, this is the first demonstration that brain iron loading influences the expression of a suite of genes causatively linked to NBIA, as well as the first demonstration of specific involvement of myelin-related gene systems.Citation4 Here we provide further evidence for involvement of myelin-related systems in mouse and human brain iron dyshomeostasis, with expanded literature and array data mining revealing 17 new myelin-related genes showing altered expression in the mouse model and 74 new myelin-related genes showing altered expression in the human NBIA brain. Some of these genes are causally linked to rare myelin diseases in which iron has not previously been implicated. We also explore the possibility that perturbation of normal relationships between iron and myelin may be a shared feature both of several groups of rare brain diseases and of various more common disorders, including the iron overload disease hemochromatosis, the myelin disease multiple sclerosis and psychiatric conditions such as major depressive disorder, bipolar disorder, autism and schizophrenia.
Methods
Animals, tissue collection and iron staining
Mice with homologous recombinant knockout of the Hfe gene and mice with the Y245X nonsense mutation in the Tfr2 gene (both on AKR background) were cross-bred to generate Hfe−/−xTfr2mut mice, as described previously.Citation3 Male wildtype mice were maintained on a standard diet containing ∼0.02% iron. To maximize iron status, male Hfe−/−xTfr2mut mice were fed an iron-supplemented diet containing 2% iron for 3 weeks before sacrifice at 3 months of age; as previously reported, dietary iron supplementation by this regimen does not increase brain iron levels in wildtype mice.Citation5 Iron in brain sections was detected by 3,3′-diaminobenzidine (DAB)-enhanced Perls' stain.Citation4 All protocols were approved by the Animal Ethics Committees of the University of Western Australia and the University of Sydney.
Gene expression measurement and analysis
Total RNA isolation, labeling, amplification and microarray probe hybridization were performed using previously described methodsCitation6,7 on brain homogenates from Hfe−/−xTfr2mut mice and wildtype mice (n ≥ 4 ) and on post-mortem basal ganglia of 2 clinicopathologically confirmed NBIA cases of unknown genetic subtype (male 66 years and female 81 years), from the Canadian Brain Tissue Bank, University of Toronto, and 2 age and gender-matched adults with no diagnosed neurological conditions from the Newcastle Brain Tissue Resource, Newcastle University, UK.Citation4 Sentrix MouseRef-8 v2 Expression BeadChips or HumanHT-12 v4 Expression BeadChips (Illumina) were used to assess gene expression, with data pre-processing, background subtraction, normalization and differential expression analysis as detailed elsewhere.Citation4,8 Tissue was obtained with fully informed consent and the study approved by the Human Research Ethics Committee of the University of Newcastle, Australia (H-2010-1219).
An expanded list of myelin-related genes was generated by more extensive literature searching and application of an augmented suite of bioinformatics tools including DAVID (http://david.abcc.ncifcrf.gov/home.jsp), KEGG (http://www.genome.jp/kegg/), NCBI Biosystems (http://www.ncbi.nlm.nih.gov/biosystems/) and AmiGO2 (http://amigo.geneontology.org/amigo). This list was compared to the differentially expressed genes from the Hfe−/−xTfr2mut brain, the NBIA patient brains and 2 modules of genes showing expression correlations in normal human basal ganglia and enriched for both NBIA-related genes and genes related to myelin and oligodendrocytes, constructed from 101 normal human brains as described previously.Citation4,9
Results
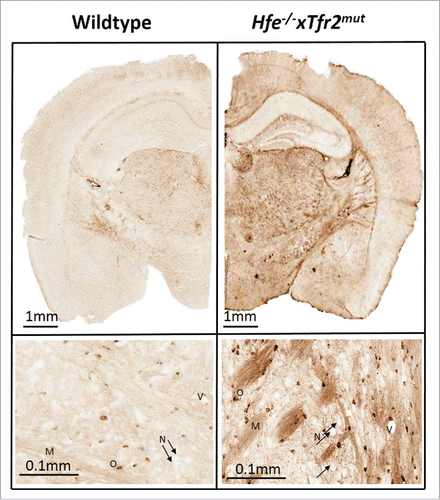
As already reported, inductively coupled plasma atomic emission spectroscopy (ICP-AES) showed that whole brain homogenates from Hfe−/−xTfr2mut mice at 3 months of age contained higher levels of iron than gender- and age-matched wildtype controls.Citation4 This was confirmed by Perls' staining, which also revealed similar anatomical patterns of heterogeneous iron distribution in Hfe−/−xTfr2mut mice fed a high iron diet to those seen in wildtype mice on a standard diet, with iron predominantly localized to myelinated structures and myelin-associated cells and with particularly high levels in basal ganglia ().
Figure 1. Iron labeling by DAB-enhanced Perls' staining in wildtype and Hfe−/−xTfr2mut mouse brain at 3 months of age. Iron is mainly accumulated in myelinated structures and associated cells. Myelin (M), oligodendrocyte (O), blood vessel (V) and black arrows show neurons (N) which do not accumulate appreciable iron even in the Hfe−/−xTfr2mut mouse brain.
We investigated an expanded list of putative myelin-related genes (n = 172), as detailed in Methods. This new list includes all but one of the published NBIA genes. Genes that were not considered strongly myelin-related in the previous paper but which have now been included in the new list based on the existence of at least 2 independent reports of oligodendrocyte or myelin-related effects are Pank2,Citation10,11 Cp,Citation12,13 Dcaf17Citation14,15 and Wdr45,Citation16,17 although not all patients with each of these mutations invariably have evidence of myelin effects.Citation4 Coasy has not been included but this may change in the future as patient series with post-mortem data are reported.
We also noted that a putative eleventh NBIA gene, RALBP1 associated Eps domain containing 1 (REPS1), was reported in the conference proceedings of the American Society of Human Genetics meeting, Boston, US, 2013.Citation18 Our expression analysis found that transcript levels of this gene are unchanged in either the Hfe−/−xTfr2mut mice (p > 0.05) or in the 2 human NBIA basal ganglia analyzed in our study. There so far appears to be no evidence for relationships with myelin or iron but little is yet known about this protein. We did not include this gene in the full analysis as it is not yet a confirmed NBIA gene.
Of the expanded list of 172 genes, the array did not contain specific probes for 35 genes and a further 30 genes failed to pass the detection threshold (p > 0.01) in both Hfe−/−xTfr2mut and wildtype mice. Of the remaining 107 genes, 40 genes (∼37%) were differentially expressed in the brain of Hfe−/−xTfr2mut mice compared with wildtype mice (p≤0 .05). This suggests the myelin transcriptome is specifically affected in these mice, since this proportion is significantly higher (Chi square p<0.0001) than that for the full transcriptome, with ∼21% of the total number of expressed genes (n = 12,318) showing altered transcript levels in the brain of the Hfe−/−xTfr2mut mice compared to wildtype mice.
Of the 40 differentially expressed myelin-related genes, 21 myelin-related genes were identified in our earlier work (comprising 5 NBIA genes and 16 other myelin-related genes), all having decreased transcript levels in Hfe−/−xTfr2mut brain except ferritin, for which transcripts were unchanged but protein levels increased.Citation4 Of the 19 newly identified genes, all but 3 also showed decreased expression (). The proportion of myelin-related genes with decreased transcript levels in Hfe−/−xTfr2mut brain (90%) is significantly higher (Chi square p < 0.0015) than that for the brain transcriptome overall (∼66%). The list includes genes encoding prominent myelin components such as myelin-associated glycoprotein (Mag) (MIM159460) and myelin oligodendrocyte glycoprotein (Mog) (MIM159465) as well as genes involved in myelin biogenesis, regulation and other relevant functions (For the full list of all 40 myelin-related genes identified in the mouse model see ; for references for each gene see Table S1).
Table 1. Myelin-related genes identified as differentially expressed in i. the Hfe−/−xTfr2mut mice in comparison with wildtype mice and ii. NBIA post-mortem basal ganglia samples compared to neurologically healthy control samples. FC: fold change. Asterisks indicate genes that were not reported as myelin-related genes in our previous paper.
We investigated the concordance of the set of 40 myelin-related genes from our mouse model of brain iron loading and a set of myelin-related genes co-expressed with NBIA-related genes in normal human brain. Two normal human basal ganglia gene co-expression modules enriched for both NBIA-related genes and genes related to myelin and oligodendrocytes were generated from analyses of 101 human brains.Citation4,9 In total these modules contained 17 (∼42%) of the 40 genes (ATP13A2, BACE1, C19orf12, CLDN11, CNP, FA2H, H6PD, MAG, MOG, MTMR2, PSAP, RTN4, SGPP2, SLC12A6, SMPD1, SOD1, TF).
Post-mortem NBIA brain samples are difficult to obtain but we have previously performed gene expression arrays on basal ganglia tissue from 2 clinicopathologically confirmed NBIA patients and matched controls.Citation4 To examine whether myelin-related gene transcript levels are selectively affected in human NBIA patients, we compared this human NBIA dataset with the full list of 172 myelin-related genes (comprising the 107 genes expressed in the mouse brain as well as those myelin-related genes either not expressed or not represented on the mouse array). Of these 172 genes, 136 were represented on the human array (DCAF17 being one notable exception), of which 91 were expressed in the basal ganglia (Cp expression was not detected). Of these, 74 (∼81%) had putatively altered transcript levels in one or more of the NBIA cases compared to controls, with 49 (∼54%) of these having putatively altered transcript levels in both NBIA patients, though not always in the same direction (for the full list of 74 genes see Table S2). This suggests the myelin transcriptome may be specifically affected in at least some NBIA patients, since the proportion of myelin-related genes with altered expression in one or more of the NBIA cases (∼81%) was significantly higher (Chi square p < 0.0001) than that for the full transcriptome, with ∼55% of all genes expressed in the basal ganglia and detected by the array. Similarly the proportion of myelin-related genes with altered expression in both NBIA cases (∼54%) was significantly higher (Chi square p < 0.0004) than that for the full transcriptome, with ∼36% of all genes expressed in the basal ganglia and detected by the array.
Myelin-related genes putatively affected in NBIA basal ganglia include important structural components of myelin e.g. MBP (MIM159430), PLP1 (MIM300401), MAG, MOG and MOBP (MIM600948), as well as genes involved in myelin or oligodendrocyte regulation such as MYT1 (MIM600379), LINGO1 (MIM609791), ASCL1 (MIM100790), RNF10 (MIM615998) and TNFRSF21 (MIM605732), encoding death receptor 6. As detailed in , of the 40 myelin-related genes with altered expression in the mouse brain, 17 (∼42%) are among the 74 myelin-related genes with putatively altered expression in the NBIA brains. Various other important myelin-related genes with putative expression changes in NBIA humans but not mouse are listed in .
Table 2. Some important myelin-related genes identified as differentially expressed in NBIA post-mortem basal ganglia samples compared to neurologically healthy control samples (asterisks indicate newly identified genes).
Discussion
The new findings support our contention that supranormal iron accumulation in myelinated structures and oligodendrocytes substantially modifies the myelin-related transcriptome, altering transcript levels for a range of disease-associated genes and potentially affecting numerous important myelin-related molecular systems. As noted elsewhere,Citation4 we attribute this to brain iron accumulation rather than other changes caused by either of the 2 mutated genes, since these responses were not observed in the single mutant mouse models used to generate the Hfe−/−xTfr2mut mice, neither of which show increased brain iron levels despite elevated liver and blood iron.
The findings suggest a close, 2-way interrelationship between iron and myelin. We hypothesize first that perturbation of myelin systems is among the initial consequences of brain iron dyshomeostasis, occurring at the very earliest stages of pre-clinical disease, and second that pathology may escalate in patients unable to mount adequate compensatory responses, with most myelin systems being affected by end-stage disease. Conversely we hypothesize that mutations in some myelin-related genes, notably various NBIA genes, will directly or indirectly reduce the iron storage capacity of myelin, resulting in abnormal iron deposition or lability.
Notwithstanding this mutual interdependence, excessive iron loading is likely to have different consequences from mutations in myelin-related genes. Mutation of any one myelin-related gene will not inevitably affect the capacity of myelin to store iron, even if the mutation causes clinical sequelae involving other functions such as nerve conduction or axon protection. Even when a single mutation has the potential to affect iron storage capacity, the phenotypic consequences may differ between patients depending on individual variations in any of a number of potential exacerbating or compensating factors involving other myelin or iron regulatory systems. This may explain why brain iron accumulation is not always present in patients with NBIA mutations.
In contrast, our results suggest that excessive increases in iron within myelinated structures and associated cells will eventually lead to widespread downstream consequences in numerous myelin systems, including transcription changes of multiple myelin-related genes. Our analyses are also revealing unexpected connections with another set of rare diseases involving myelin but not, as far as we know, previously recognized to involve iron. Notably we observed decreased transcript levels in both Hfe−/−xTfr2mut mouse and at least one human NBIA case for 2 genes encoding proteins with different functions yet both causatively linked to very rare hypomyelinating leukodystrophies, the heat shock protein 60 HSPd1 gene, linked to mitochondrial Hsp60 chaperonopathy (MIM612233), and the gap junction GJC2 gene, linked to both Pelizaeus-Merzbacher-like disease (MIM608804) and a mild, late-onset, slowly progressive, complicated spastic paraplegia (MIM613206). Decreased transcript levels may give rise to partial loss-of-function phenotypes but further research is needed to determine the effects, if any, in our model.
Regarding spastic paraplegias and other peripheral diseases, we focused on genes considered primarily relevant to central nervous system (CNS) myelin but also noted changes in transcript levels for various genes important for peripheral myelin, including peripheral myelin protein 2 Pmp2 (MIM170715), annexin Anxa2 (MIM151740), Gdap1 (MIM606598) and Mtmr2 (MIM603557), both linked to Charcot-Marie-Tooth peripheral neuropathies. Systematic investigation will probably reveal more such genes in the future. Few if any investigations have applied modern iron staining techniques in the peripheral nervous system in such diseases or in the common iron disorder hemochromatosis but a 2010 study reported a high rate (26%) of idiopathic polyneuropathy in a small clinical series of hemochromatosis patients (n = 46).Citation19
The large number of myelin-related genes with altered transcript levels is perhaps more indicative of generalized actions of iron on myelin per se rather than specific effects of iron loading on the transcript levels of individual genes. As transcript levels were almost invariably decreased in the mouse model, one possibility is that excess iron leads to myelin degradation, causing a gross decrease in total brain myelin and proportionate decreases of myelin transcripts relative to total brain transcripts. However, as we reported earlier, semi-quantitative analyses revealed no apparent differences in myelination by electron microscopy or in gross myelin content by Luxol fast blue staining and there were no transcript level changes for 2 major myelin structural components, MBP and PLP.Citation4 Instead there may be more subtle adjustments in myelin structural and regulatory subsystems as the amount of iron stored in myelin starts to ramp up beyond normal levels. For example, some of the transcripts showing changes in Hfe−/−xTfr2mut mouse or humans, such as Opalin (Gene ID 93377), Mtmr2 and Anxa2, encode genes important in myelin paranodal regions and may have subtle effects on the nodes of Ranvier and nerve conduction properties or on the axon without affecting myelin quantity or gross integrity. In addition, altered expression of some neuronal genes may cause myelin problems. For example, the SLC12A6 gene, which shows decreased expression in the Hfe−/−xTfr2mut mouse brain, codes for the neuronal potassium- and chloride-cotransporter 3 (KCC3).Citation20 Mutations in SLC12A6 are associated with hereditary motor and sensory neuropathy with agenesis of corpus callosum (HMSN/ACC), with axonal swelling and myelin impairment.Citation21 Dysfunction of this transporter therefore appears to affect the axonal ion-fluid osmotic equilibrium, resulting in local edema and potentially indirectly affecting myelin.
In this regard it is worth noting that various genes identified in our analyses have roles beyond their myelin-related functions. As just one example, the Klotho (Kl) gene (MIM604824) is implicated in major myelin pathways, with null mutant mice showing impaired myelination, but has also been reported to confer resistance to oxidative stressCitation22 and to protect hippocampal and dopaminergic neurons from neurodegeneration by regulating redox systems,Citation23,24 as well as having roles in suppression of aging.Citation25,26 The reduced expression of Klotho in the Hfe−/−xTfr2mut mice might therefore confer increased vulnerability to various degenerative changes, whether involving myelin, neurons or other brain systems.
The hemochromatosis modeled by the Hfe−/−xTfr2mut mouse would usually be considered likely to be far less severe than most NBIAs, both with regard to putative brain iron abnormalities as well as with regard to the severity and progression rate of clinical features. We therefore believe that, with respect to brain iron loading, this new model provides insights into early perturbations in molecular systems during a period corresponding to a pre-clinical disease stage in which compensatory responses are still sufficient to maintain the overall homeostasis of myelin and iron-related systems. For example, evidence was found for reductions in Lingo1 expression; this might enhance myelination, with antibodies to LINGO1 increasing myelin thickness in early clinical trials.Citation27
The high concordance of the expanded gene list with the 2 human post-mortem NBIA basal ganglia samples substantiates the clinical relevance of our observations and testifies to myelin abnormalities in at least some cases of end-stage NBIA. The direction of changes often differed between the 2 NBIA samples but numerous transcripts appeared affected in each, suggesting that even though the precise nature of the changes might differ, myelin systems were likely to be extensively perturbed in both. As reviewed elsewhere,Citation28 MBP appears to be the only myelin protein required for myelin growth; myelin assembly can occur in the complete absence of other myelin proteins including PLP, MOG, CLDN11 (MIM601326), MAL and PMP22 (MIM601097). Notably both samples showed putative increases in MBP transcript levels, suggesting the possibility of ongoing attempts to enhance myelin production. This could serve to either increase iron reservoir capacity by increasing myelin levels above normal or repair myelin damage. While we have so far found no evidence for clear myelin damage by light or electron microscopy, the second possibility is perhaps more likely based on current evidence since, as detailed in our earlier paper, there appears to be no gross significant change in myelin levels as detected by Luxol fast blue staining.
However the NBIAs exhibit considerable heterogeneity. Difficulties in obtaining samples due to the extreme rarity of NBIA cases limits the inferences we can confidently draw and we have been unable to attempt validation of the observations statistically or at the protein level. In addition our samples were from elderly patients of unknown genetic etiology who may differ considerably from patients having more severe NBIAs with childhood onset. For more complete understanding, including assessment of whether myelin-related changes are specifically associated with one or more NBIA subtypes rather than non-specific age-related brain degenerative changes, it will be important to conduct future analyses using both arrays and other approaches on greater numbers of samples, including some more relevant to childhood NBIA.
One of the main challenges in these analyses has been the lack of a comprehensive, evidence-based list of myelin-related genes, including genes encoding all the protein components of myelin, genes encoding enzymes involved in synthesis of myelin lipids and other myelin components, regulatory genes, for example encoding oligodendrocyte and myelin-related transcription factors, and genes with known links to myelin diseases. Despite our efforts to address this, there are other relevant genes not covered by our analyses. For example we have not performed an extensive search for genes involved in peripheral myelin. It is also possible that some genes may need to be removed from the list as new evidence comes to light, although we have attempted to restrict inclusion to genes for which the evidence for relationships with myelin is relatively strong e.g., myelin abnormalities in patients with mutations or genetically modified mouse models.
It will be of interest in the future to investigate whether any of the 172 myelin-related genes are causal genes for NBIA using exome sequencing or other sequencing approaches. The most obvious candidates to consider first would be those with the greatest expression changes, notably MOBP, MBP and PLP. Mutations in MOBP and MBP do not appear to have clear human disease phenotypes but, as reviewed elsewhere,Citation29 various MRI studies have reported changes consistent with brain iron abnormalities in patients with Pelizaeus-Merzbacher disease, a myelin disease caused by mutations in PLP. However post-mortem histopathological studies have yet to be done to confirm these findings and it is unclear if any such iron abnormalities are also accompanied by neurodegeneration. Findings from Plp deficient mice have shown neurodegeneration in the form of axonal degeneration but brain iron levels were not investigated in these models.Citation30
It is often speculated that excess iron is important in the pathogenesis of neurodegenerative conditions such as Alzheimer's disease but, despite involvement of NBIA genes, we see little evidence for neurodegenerative disease, with neurons spared even in states of substantial brain iron loading (see ). Instead changes primarily involve myelin. Iron has sometimes been proposed to be a bystander in NBIA and while myelin pathology has been reported in a subset of NBIA patients, it is not usually considered a central feature of the disease process. Our data provide strong evidence that disruption of iron-myelin homeostasis is a prominent and common mechanistic feature underlying most, if not all, NBIAs. The results also unexpectedly implicate iron in several rare myelin disorders and also appear likely to provide fresh insights into several clinically diverse and far more prevalent conditions including multiple sclerosis, hemochromatosis and various common psychiatric disorders.
As noted in our earlier paper,Citation4 interrelationships between iron and myelin are rarely considered in this context, although both have been separately proposed to be contribute to depression, bipolar disorder, autism, schizophrenia and other psychiatric conditions. As we also detailed previously,Citation4 there have also been thought-provoking proposals that increased white matter iron, demyelination and axonal degeneration may drive the shift from presymptomatic to clinical Huntington's disease and development of psychiatric symptoms.
Venesection or iron chelation is used to treat hemochromatosis and iron chelators are currently being trialed in NBIA patients. These approaches, together with myelin-based treatments, may provide simple new strategies for managing these as well as other disorders involving iron and myelin.
Abbreviations
| CNS | = | central nervous system |
| DAB | = | 3,3′-diaminobenzidine |
| FC | = | fold change |
| MIM | = | Mendelian Inheritance in Man |
| NBIA | = | neurodegeneration with brain iron accumulation |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KRAD_S_1198458.docx
Download MS Word (110.3 KB)Acknowledgments
The authors would like to thank the UK Brain Expression Consortium affiliated with the UCL Institute of Neurology, King's College London, Istituto di Ricerca Genetica e Biomedica, CNR, Cagliari and the University of Edinburgh; LN Hazrati for assistance with provision of tissue from the Canadian Brain Tissue Bank (University of Toronto, Canada) and the Newcastle Brain Tissue Resource, supported by the MRC, the Alzheimer Research Trust and Alzheimer Society through the Brains for Dementia Research Initiative and NIHR Biomedical Research Center grants.
Funding
This study was supported by the National Health and Medical Research Council of Australia (572601, 1042370, 1020437, 1078747), Fremantle Hospital Medical Research Foundation, Australian Society for Medical Research, EPSRC (EP/D066654/1), the UK Medical Research Council (MR/J004758/1) and the Wellcome Trust (WT104033/Z/14/Z).
References
- Gregory A, Hayflick S. Neurodegeneration with Brain Iron Accumulation Disorders Overview. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, eds. GeneReviews(R). Seattle WA: University of Washington, Seattle, 1993
- Johnstone D, Milward EA. Molecular genetic approaches to understanding the roles and regulation of iron in brain health and disease. J Neurochem 2010; 113:1387-402; PMID:20345752
- Delima RD, Chua AC, Tirnitz-Parker JE, Gan EK, Croft KD, Graham RM, Olynyk JK, Trinder D. Disruption of hemochromatosis protein and transferrin receptor 2 causes iron-induced liver injury in mice. Hepatology 2012; 56:585-93; PMID:22383097; http://dx.doi.org/10.1002/hep.25689
- Heidari M, Johnstone DM, Bassett B, Graham RM, Chua ACG, House MJ, Collingwood JF, Bettencourt C, Houlden H, Ryten M, et al. Brain iron accumulation affects myelin-related molecular systems implicated in a rare neurogenetic disease family with neuropsychiatric features. Mol Psychiatry 2016; PMID:26728570; http://www.nature.com/mp/journal/vaop/ncurrent/full/mp2015192a.html
- Johnstone D, Milward EA. Genome-wide microarray analysis of brain gene expression in mice on a short-term high iron diet. Neurochem Int 2010; 56:856-63; PMID:20350576; http://dx.doi.org/10.1016/j.neuint.2010.03.015
- Acikyol B, Graham RM, Trinder D, House MJ, Olynyk JK, Scott RJ, Milward EA, Johnstone DM. Brain transcriptome perturbations in the transferrin receptor 2 mutant mouse support the case for brain changes in iron loading disorders, including effects relating to long-term depression and long-term potentiation. Neuroscience 2013; 235:119-28; PMID:23333676; http://dx.doi.org/10.1016/j.neuroscience.2013.01.014
- Johnstone D, Graham RM, Trinder D, Delima RD, Riveros C, Olynyk JK, Scott RJ, Moscato P, Milward EA. Brain transcriptome perturbations in the Hfe(−/−) mouse model of genetic iron loading. Brain Res 2012; 1448:144-52; PMID:22370144; http://dx.doi.org/10.1016/j.brainres.2012.02.006
- Johnstone D, Riveros C, Heidari M, Graham R, Trinder D, Berretta R, Olynyk J, Scott R, Moscato P, Milward E. Evaluation of Different Normalization and Analysis Procedures for Illumina Gene Expression Microarray Data Involving Small Changes. Microarray. 2013; 2:131-52; http://www.mdpi.com/2076-3905/2/2/131
- Bettencourt C, Forabosco P, Wiethoff S, Heidari M, Johnstone DM, Botia JA, Collingwood JF, Hardy J, Milward EA, Ryten M, et al. Gene co-expression networks shed light into diseases of brain iron accumulation. Neurobiol Dis 2016; 87:59-68; PMID:26707700; http://dx.doi.org/10.1016/j.nbd.2015.12.004
- Bhatia KP, Schneider S. Metal Related Neurodegenerative Disease. Int Rev Neurobiol. London, UK: Academic Press, 2013; 434
- Li A, Paudel R, Johnson R, Courtney R, Lees AJ, Holton JL, Hardy J, Revesz T, Houlden H. Pantothenate kinase-associated neurodegeneration is not a synucleinopathy. Neuropathol Appl Neurobiol 2013; 39:121-31; PMID:22416811; http://dx.doi.org/10.1111/j.1365-2990.2012.01269.x
- Zhao L, Hadziahmetovic M, Wang C, Xu X, Song Y, Jinnah HA, Wodzinska J, Iacovelli J, Wolkow N, Krajacic P, et al. Cp/Heph mutant mice have iron-induced neurodegeneration diminished by deferiprone. J Neurochem 2015; 135(5):958-74
- Schulz K, Vulpe CD, Harris LZ, David S. Iron efflux from oligodendrocytes is differentially regulated in gray and white matter. J Neurosci 2011; 31:13301-11; PMID:21917813; http://dx.doi.org/10.1523/JNEUROSCI.2838-11.2011
- Alazami AM, Schneider SA, Bonneau D, Pasquier L, Carecchio M, Kojovic M, Steindl K, de Kerdanet M, Nezarati MM, Bhatia KP, et al. C2orf37 mutational spectrum in Woodhouse-Sakati syndrome patients. Clin Genet 2010; 78:585-90; PMID:20507343; http://dx.doi.org/10.1111/j.1399-0004.2010.01441.x
- Ben-Omran T, Ali R, Almureikhi M, Alameer S, Al-Saffar M, Walsh CA, Felie JM, Teebi A. Phenotypic heterogeneity in Woodhouse-Sakati syndrome: two new families with a mutation in the C2orf37 gene. Am J Med Genet A 2011; 155A:2647-53; PMID:21964978; http://dx.doi.org/10.1002/ajmg.a.34219
- Okamoto N, Ikeda T, Hasegawa T, Yamamoto Y, Kawato K, Komoto T, Imoto I. Early manifestations of BPAN in a pediatric patient. Am J Med Genet A 2014; 164:3095-9; http://dx.doi.org/10.1002/ajmg.a.36779
- Rathore GS, Schaaf CP, Stocco AJ. Novel mutation of the WDR45 gene causing β-propeller protein-associated neurodegeneration. Mov Disord 2014; 29:574-5; PMID:24610255; http://dx.doi.org/10.1002/mds.25868
- Drecourt A, Boddaert N, Desguerre I, Chretien D, Munnich A, Rotig A. REPS1 is a novel gene of Neurodegeneration with Brain Iron Accumulation. American Society of Human Genetics meeting. Boston, US, 2013
- Wouthuis SF, van Deursen CT, te Lintelo MP, Rozeman CA, Beekman R. Neuromuscular manifestations in hereditary haemochromatosis. J Neurol 2010; 257:1465-72; PMID:20358215; http://dx.doi.org/10.1007/s00415-010-5548-x
- Shen MR, Chou CY, Hsu KF, Liu HS, Dunham PB, Holtzman EJ, Ellory JC. The KCl cotransporter isoform KCC3 can play an important role in cell growth regulation. Proc Natl Acad Sci U S A 2001; 98:14714-9; PMID:11724933; http://dx.doi.org/10.1073/pnas.251388798
- Dupre N, Howard HC, Rouleau GA. Hereditary Motor and Sensory Neuropathy with Agenesis of the Corpus Callosum. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, et al., eds. GeneReviews(R). Seattle University of Washington, 1993
- Chateau MT, Araiz C, Descamps S, Galas S. Klotho interferes with a novel FGF-signalling pathway and insulin/Igf-like signalling to improve longevity and stress resistance in Caenorhabditis elegans. Aging 2010; 2:567-81; PMID:20844315; http://dx.doi.org/10.18632/aging.100195
- Brobey RK, German D, Sonsalla PK, Gurnani P, Pastor J, Hsieh CC, Papaconstantinou J, Foster PP, Kuro-o M, Rosenblatt KP. Klotho Protects Dopaminergic Neuron Oxidant-Induced Degeneration by Modulating ASK1 and p38 MAPK Signaling Pathways. PloS one 2015; 10:e0139914; PMID:26452228; http://dx.doi.org/10.1371/journal.pone.0139914
- Zeldich E, Chen CD, Colvin TA, Bove-Fenderson EA, Liang J, Tucker Zhou TB, Harris DA, Abraham CR. The neuroprotective effect of Klotho is mediated via regulation of members of the redox system. J Biol Chem 2014; 289:24700-15; PMID:25037225; http://dx.doi.org/10.1074/jbc.M114.567321
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997; 390:45-51; PMID:9363890; http://dx.doi.org/10.1038/36285
- Razzaque MS, Sitara D, Taguchi T, St-Arnaud R, Lanske B. Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. FASEB J 2006; 20:720-2; PMID:16436465
- Sun JJ, Ren QG, Xu L, Zhang ZJ. LINGO-1 antibody ameliorates myelin impairment and spatial memory deficits in experimental autoimmune encephalomyelitis mice. Sci Rep 2015; 5:14235; PMID:26383267; http://dx.doi.org/10.1038/srep14235
- Saher G, Brugger B, Lappe-Siefke C, Mobius W, Tozawa R, Wehr MC, Wieland F, Ishibashi S, Nave KA. High cholesterol level is essential for myelin membrane growth. Nat Neurosci 2005; 8:468-75; PMID:15793579
- Stevenson RE, Tarpey P, May MM, Stratton MR, Schwartz CE. Arena syndrome is caused by a missense mutation in PLP1. Am J Med Genet A 2009; 149A:1081; PMID:19396823; http://dx.doi.org/10.1002/ajmg.a.32795
- Woodward K, Malcolm S. Proteolipid protein gene: Pelizaeus–Merzbacher disease in humans and neurodegeneration in mice. Trends Genet 1999; 15:125-8; PMID:10203813; http://dx.doi.org/10.1016/S0168-9525(99)01716-3