
Abstract
Amyotrophic lateral sclerosis (ALS) is an invariably fatal adult-onset neurodegenerative disorder; approximately 10% of ALS is monogenic but all ALS exhibits significant heritability. The skeletal muscle sodium channelopathies are a group of inherited, non-dystrophic ion channel disorders caused by heterozygous point mutations in the SCN4A gene, leading to clinical manifestations of congenital myotonia, paramyotonia, and periodic paralysis syndromes. We provide clinical and genetic evidence of concurrence of these two rare disorders which implies a possible shared underlying pathophysiology in two patients. We then identify an enrichment of ALS-associated mutations in another sodium channel, SCN7A, from whole genome sequencing data of 4495 ALS patients and 1925 controls passing multiple testing correction (67 variants, p = 0.0002, Firth logistic regression). These findings suggest dysfunctional sodium channels may play a role upstream in the pathogenesis of ALS in a subset of patients, potentially opening the door to novel personalized medicine approaches.
Keywords:
Case reports
We present a 72-year-old Caucasian gentleman who developed ALS on a background of sodium channel myotonia due to p.Ser1159Pro mutation in SCN4A (). This mutation co-segregated with myotonia in the index pedigree () and it is absent from 141,456 control individuals within gnomAD (Citation1). Within the SC4A protein p.Ser1159Pro is located in the S4-S5 loop in close proximity to previously described p.Ala1152Asp, p.Ala1156Thr, and p.Ile1160Val mutations, and it is predicted to be pathogenic in silico (Citation2). The patient reported lifelong mild cramping and myotonia affecting his hands and eyelids. At age of 70, he presented with progressive limb weakness initially most prominent in the left hand followed by bulbar weakness. Clinical examination revealed weakness and wasting in all limbs, increased muscle tone, brisk tendon reflexes, and widespread fasciculations. Tongue movements were slow and spastic, and marked dysarthria was noted. Electrophysiological studies revealed chronic neurogenic changes on a background of widespread fibrillation and fasciculation, but also sharp waves and myotonia. A clinical diagnosis of ALS was made. Lumbar puncture, MRI brain and spine imaging, and blood investigations did not demonstrate an alternative cause. He commenced Riluzole, but deteriorated and died from respiratory infection 2 years after the onset of ALS symptoms.
Figure 1 Schematic representation of SCN4A mutations identified in ALS patients. Clinically reported neuromuscular phenotype highlighted in boxes. Identified genetic changes within SCN4A are proposed to lead to motor neuron toxicity by one of two mechanisms: either directly via excessive membrane sodium permeability leading to hyperexcitability and ultimately excitotoxicity; or indirectly via muscle hyperexcitability leading to retrograde motor neuron toxicity.
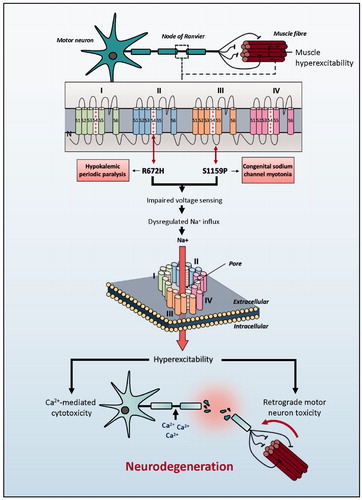
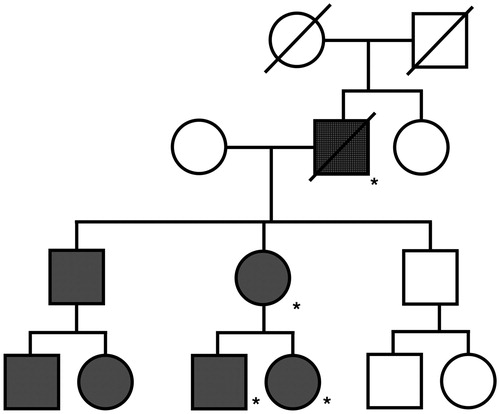
Figure 2 Pedigree from index case. Pedigree showing co-segregation of SCN4A p.Ser1159Pro with congenital myotonia. (shaded: congenital myotonia, crosshatch: ALS and congenital myotonia, * = SCN4A p.Ser1159Pro mutation confirmed).
We hypothesized that SCN4A mutation may have predisposed our patient to develop ALS. We, therefore, screened 1138 familial ALS patients (als.umassmed.edu/) for additional pathogenic mutations within SCN4A. While no further patients carrying p.Ser1159Pro mutations were identified, we identified a single Caucasian male carrying a rare p.Arg672His mutation (ClinVar: www.ncbi.nlm.nih.gov/clinvar/variation/VCV000005912.1, ) who developed dysarthria and dysphagia aged 66 years. A diagnosis of familial ALS was made 12 months later. No other mutations in previously described ALS genes were identified. Despite Riluzole, he died from respiratory failure 18 months after symptom onset. p.Arg672His is typically associated with hypokalemic periodic paralysis (hypoPP) type 2, which may clinically overlap with sodium channel myotonia (Citation3). Available records were insufficient to confirm clinical/electrophysiological evidence of sodium channel dysfunction.
Discussion
We are the first to report clinical and genetic evidence of SCN4A-channelopathy preceding development of ALS. Observed concurrence may be coincidence, but both conditions are rare (Citation4) and, therefore, a common pathway for pathogenesis should be considered. Extensive observational evidence has linked ALS to sodium channel dysfunction, but our genetic data tentatively places sodium channel dysfunction upstream in the development of ALS in selected cases.
While the pathophysiology of ALS is not fully elucidated, aberrant sodium conductance is implicated in both sporadic and familial forms (reviewed in (Citation5)) with excitotoxicity considered responsible for motor neuron death. NaV1.4 (encoded by SCN4A) is a voltage-gated sodium channel expressed predominantly in muscle sarcolemma. Changes in muscle excitability have been linked to retrograde motor neuron toxicity (Citation6) () but it is noticeable that SCN4A is expressed within the CNS (Citation7,Citation8).
The two SCN4A mutations we have identified are typically associated with opposite effects on muscle excitability – hyperexcitability and sodium channel myotonia (p.Ser1159Pro) versus hypoexcitability and hypoPP type 2 (p.Arg672His) (Citation3). p.Arg672His enables an abnormal inward cation “gating pore” current which depolarizes the cell leading to inactivation of sodium channels and hypoexcitability; however, similar mutations are linked to paradoxical depolarization at low-normal extracellular [K+] leading to hyperexcitability (Citation9). This explains the clinical overlap with sodium channel myotonia (Citation3). We hypothesize that p.Arg672His-SCN4A would produce membrane hyperexcitability in the CNS where extracellular [K+] is lower (Citation10). In both of our patients, we speculate that abnormal NaV1.4 channels within motor neuron membranes predisposed to depolarization-induced cellular excitotoxicity, leading to the development of ALS (). Alternatively, changes in muscle excitability have been linked to retrograde motor neuron toxicity, and may have played a role in the etiology of these cases () (Citation6). Further mechanistic studies are required.
We hypothesized that ALS may be linked to genetic mutations in other voltage-gated ion channels. Rare-variant burden testing using whole genome sequencing data from 4495 ALS patients and 1925 controls (databrowser.projectmine.com/) within the superfamily of voltage-gated ion channels (Accession:ssf81324) identified one gene, SCN7A, which passed multiple testing correction (p = 0.00029, beta = 0.41, Firth logistic regression; rare-variants defined as missense and MAF < 0.01, Supplementary Table) consistent with an enrichment of ALS-associated mutations. SCN7A encodes a type II sodium channel, NaX, and is expressed in glial cells. NaX is not voltage-gated, and channel permeability is proportional to extracellular [Na+] so as to mediate [Na+] homeostasis (Citation11). We identified 67 rare predicted pathogenic (Citation12) ALS-associated variants within SCN7A of which 3 are premature stop codon variants which undoubtedly lead to haploinsufficiency (Supplementary Table). We propose that SCN7A loss of function may disrupt extracellular [Na+] homeostasis and lead to neuronal hyperexcitability.
Our findings add to the growing body of evidence for sodium channel dysfunction in ALS. Identification of upstream genetic mutations may ultimately provide the basis for a personalized medicine approach.
Supplementary_Table_Revised.xlsx
Download MS Excel (15.9 KB)Acknowledgments
The authors acknowledge the Project MinE GWAS Consortium and the ALS Variant Server.
Declaration of interest
The authors declare no conflicts of interest.
Additional information
Funding
References
- Durran S. Genetic and molecular studies of skeletal muscle channelopathies. Doctoral thesis, UCL London: University of College London. 2015.
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2019;581:434–43.
- Huang S, Zhang W, Chang X, Guo J. Overlap of periodic paralysis and paramyotonia congenita caused by SCN4A gene mutations two family reports and literature review. Channels (Austin). 2019;13:110–9.
- Horga A, Raja Rayan DL, Matthews E, Sud R, Fialho D, Durran SCM, et al. Prevalence study of genetically defined skeletal muscle channelopathies in England. Neurology 2013;80:1472–5.
- Bae JS, Simon NG, Menon P, Vucic S, Kiernan MC. The puzzling case of hyperexcitability in amyotrophic lateral sclerosis. J Clin Neurol. 2013;9:65–74.
- Camerino GM, Fonzino A, Conte E, De Bellis M, Mele A, Liantonio A, et al. Elucidating the contribution of skeletal muscle ion channels to amyotrophic lateral sclerosis in search of new therapeutic options. Sci Rep. 2019;9:1–15.
- Candenas L, Seda M, Noheda P, Buschmann H, Cintado CG, Martin JD, et al. Molecular diversity of voltage-gated sodium channel alpha and beta subunit mRNAs in human tissues. Eur J Pharmacol. 2006;541:9–16.
- Duan BC, Wong LC, Lee WT. Alternating hemiplegia and paroxysmal torticollis caused by SCN4A mutation: a new phenotype? Neurology 2019;93:673–4.
- Mi W, Rybalchenko V, Cannon SC. Disrupted coupling of gating charge displacement to Na + current activation for DIIS4 mutations in hypokalemic periodic paralysis. J Gen Physiol. 2014;144:137–45.
- Kann O, Hollnagel J-O, Elzoheiry S, Schneider J. Energy and potassium ion homeostasis during gamma oscillations. Front Mol Neurosci. 2016;9:47.
- Hiyama TY, Watanabe E, Ono K, Inenaga K, Tamkun MM, Yoshida S, et al. Na(x) channel involved in CNS sodium-level sensing. Nat Neurosci. 2002;5:511–2.
- Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894.